Qu’est-ce qu’une analyse ciblée et pourquoi l’utiliser ?
Définition d’une analyse ciblée
Une analyse ciblée consiste à détecter et quantifier une ou plusieurs molécules que l’on s’attend à trouver dans l’échantillon. Contrairement à une analyse non ciblée, elle ne cherche pas à observer l’ensemble des composés présents, mais uniquement ceux qui sont définis à l’avance. Pour réaliser une analyse ciblée, il est indispensable de connaître préalablement les caractéristiques de la (ou des) molécule(s) d’intérêt, notamment :
-
le poids moléculaire ;
-
la masse sur charge (m/z) de l’ion précurseur, idéalement le plus abondant ;
-
les ions fragments caractéristiques issus de la fragmentation de cet ion précurseur ;
-
et, si le spectromètre de masse est couplé à une chromatographie (LC-MS), le temps de rétention de la molécule.
Principe de la méthode MRM / SRM
La technique de référence pour l’analyse ciblée en spectrométrie de masse est le MRM (Multiple Reaction Monitoring), également appelé SRM (Single Reaction Monitoring) les deux termes désignent fondamentalement le même principe.
Cette approche est principalement utilisée avec les spectromètres de masse à triple quadripôle.
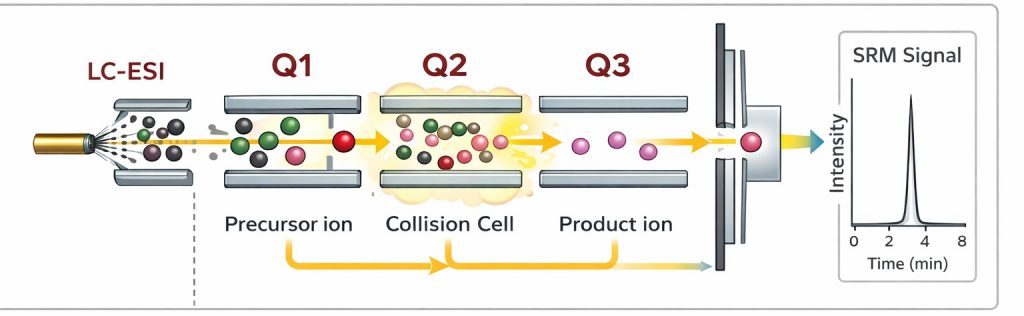
Fonctionnement d’un triple quadripôle en mode MRM
Le fonctionnement se décompose en trois étapes (figure 1) :
-
Premier quadripôle (Q1)
Il est réglé pour ne laisser passer que l’ion précurseur correspondant au m/z de la molécule d’intérêt. -
Cellule de collision (Q2)
L’ion précurseur sélectionné est fragmenté par collision avec un gaz (CID), produisant des ions fils caractéristiques. -
Troisième quadripôle (Q3)
Il filtre un ion fils spécifique à la fois.
Pour une meilleure confirmation de l’identité de la molécule, on peut surveiller deux ou trois transitions (précurseur → ions fils), mesurées à des temps différents.
Sélectivité accrue
Même si d’autres ions interférents possèdent le même m/z que l’ion précurseur et traversent Q1, ils ne génèrent généralement pas les mêmes ions fils après fragmentation.
Ils sont donc éliminés au niveau de Q3, ce qui garantit une détection très fiable et spécifique.
Gain majeur en sensibilité et réduction du bruit
L’un des principaux avantages du mode MRM est le gain considérable en sensibilité et la réduction du bruit de fond.
En mode full scan, le quadripôle balaie successivement tous les m/z d’une gamme donnée.
Par exemple :
-
un scan de m/z 1 à 1000 en 1 seconde donne environ 1 ms par ion, ce qui limite fortement la sensibilité.
En mode MRM, le quadripôle est fixé sur un m/z précis :
-
l’ion d’intérêt bénéficie alors d’un temps de résidence beaucoup plus long, ce qui améliore significativement le signal détecté.
Pourquoi l’analyse ciblée est surtout adaptée au triple quadripôle
L’analyse ciblée repose sur la sélection séquentielle d’ions, ce qui la rend particulièrement adaptée aux instruments de type quadripôle.
-
Les hexapôles ou octopoles sont possibles, mais leur sélectivité est nettement inférieure.
-
Les configurations quadripôle + piège ionique sont théoriquement intéressantes, mais :
-
l’accumulation des ions dans le piège est lente ;
-
la fragmentation repose sur des collisions multiples, plus longues ;
→ ce qui rend l’analyse ciblée peu efficace en pratique.
-
-
Les analyseurs Orbitrap, FT-ICR ou TOF, qui détectent tous les ions simultanément, ne tirent pas d’avantage particulier du MRM.
Dans ces derniers cas, on privilégie plutôt le PRM (Parallel Reaction Monitoring), qui correspond simplement à un MS/MS classique, où tous les ions fragments sont détectés en parallèle.
Simplicité d’utilisation et robustesse
Un autre avantage majeur du mode MRM sur triple quadripôle est sa simplicité d’exploitation :
-
une fois la méthode développée et validée ;
-
la détection et la quantification sont robustes et reproductibles ;
-
la méthode peut être utilisée efficacement par du personnel peu spécialisé en spectrométrie de masse.
Développement d’une méthode d’analyse ciblée
Pour paramétrer une méthode d’analyse ciblée, il est nécessaire de définir précisément les transitions MRM à surveiller.
Cela consiste, dans un premier temps, à renseigner :
-
le m/z de l’ion précurseur au niveau du premier quadripôle (Q1) ;
-
puis le m/z de l’ion fils sélectionné par le troisième quadripôle (Q3).
Dans la grande majorité des cas, le spectromètre de masse est couplé à une chromatographie (LC-MS). Il est alors indispensable de définir une fenêtre temporelle pendant laquelle le spectromètre surveille la molécule d’intérêt.
Fenêtre de temps (scheduled MRM)
Par exemple, si le temps de rétention de la molécule est de 5 minutes, on peut définir une fenêtre comprise entre 4,8 et 5,2 minutes.
La largeur de cette fenêtre dépend principalement de la largeur du pic chromatographique et de la stabilité de la rétention.
L’utilisation de fenêtres temporelles permet :
-
d’optimiser le temps passé sur chaque transition ;
-
d’augmenter la sensibilité ;
-
et de surveiller un plus grand nombre de molécules dans une même analyse.
Importance des ions fils de confirmation
Il est fortement recommandé de surveiller au moins deux ions fils pour chaque molécule :
-
un ion quantificateur (le plus intense) ;
-
un ion qualificateur pour la confirmation.
Malgré la combinaison de plusieurs critères de sélectivité (Q1, Q3 et temps de rétention), il peut exister des molécules interférentes, souvent des isomères, possédant des caractéristiques très proches de la molécule d’intérêt.
Dans ce cas, la confirmation repose sur le ratio des surfaces chromatographiques entre l’ion quantificateur et l’ion qualificateur. Ce ratio doit rester constant, quelle que soit la concentration de la molécule, car à énergie de collision constante, les intensités relatives des ions fils ne dépendent pas de la concentration. Toute variation significative de ce ratio est un indicateur potentiel d’interférence ou de problème analytique.
Temps de transition (dwell time)
Un autre paramètre clé du développement d’une méthode ciblée est le temps de transition, aussi appelé dwell time :
il correspond au temps pendant lequel une transition donnée est mesurée.
-
Si le temps de transition est trop court (quelques millisecondes), la sensibilité diminue.
-
S’il est trop long, le nombre de points acquis sur le pic chromatographique devient insuffisant, ce qui dégrade la reconstruction du pic.
Exemple illustratif
Si l’on fixe un temps de transition de 1 seconde et que la largeur du pic chromatographique est de 6 secondes, alors seulement 6 scans MS/MS seront acquis sur le pic. Ce nombre est insuffisant pour une intégration fiable.
Il n’existe pas de règle absolue, mais en pratique, une dizaine à une quinzaine de points par pic constitue un bon compromis entre sensibilité et qualité de reconstruction. Un temps de transition de 1 seconde est un cas extrême ; en pratique, des valeurs de l’ordre de 20 à 100 ms sont généralement suffisantes pour une analyse ciblée robuste.
Cas des méthodes multi-composés
Dans la plupart des applications réelles, une analyse ciblée ne porte pas sur une seule molécule, mais sur plusieurs dizaines de composés.
Dans ce cas :
-
le temps de cycle total correspond à la somme des temps de transition de toutes les transitions actives dans une même fenêtre temporelle ;
-
si plusieurs molécules ont des temps de rétention proches, le temps de cycle peut rapidement atteindre 1 seconde ou plus.
Il est donc essentiel d’optimiser :
-
le nombre de transitions par molécule ;
-
le temps de transition individuel ;
-
et les fenêtres de rétention,
afin de conserver un nombre suffisant de points par pic chromatographique tout en maximisant la sensibilité.
La quantification en analyse ciblée
La principale application de l’analyse ciblée en spectrométrie de masse est la quantification.<Pour obtenir une quantification fiable et reproductible, l’utilisation d’un étalon interne est indispensable.
Rôle de l’étalon interne
L’étalon interne permet de corriger :
-
les variations de rendement d’extraction lors de la préparation de l’échantillon ;
-
les effets de matrice;
-
les fluctuations de sensibilité du spectromètre de masse au cours de l’analyse.
La quantification repose alors sur le rapport de signal entre la molécule d’intérêt et son étalon interne, ce qui améliore fortement la précision et la justesse des résultats.
Étalons internes isotopiquement marqués
L’étalon interne idéal est l’isotope marqué de la molécule d’intérêt.
Un tel isotope présente :
-
les mêmes propriétés physico-chimiques que la molécule à doser;
-
le même comportement chromatographique ;
-
la même efficacité d’ionisation et de fragmentation.
Il subit donc exactement les mêmes étapes que la molécule d’intérêt, depuis l’extraction jusqu’à la détection, ce qui permet une normalisation optimale du signal. La seule différence entre l’isotope marqué et la molécule native est la masse, différence que le spectromètre de masse distingue sans difficulté.
Principe de la quantification
En pratique :
-
une quantité connue d’étalon interne est ajoutée à chaque échantillon ;
-
le signal de la molécule d’intérêt est rapporté à celui de l’étalon interne ;
-
la concentration est déterminée à partir d’une courbe d’étalonnage construite sur ce rapport de signaux.
Cette approche constitue aujourd’hui le standard de référence pour la quantification en LC-MS/MS en mode MRM.
Analyse ciblée en protéomique
L’analyse ciblée est historiquement et majoritairement utilisée en métabolomique,[1] pour une raison principale :
les standards analytiques des molécules d’intérêt sont généralement disponibles sous forme pure. Cela permet, de déterminer facilement le temps de rétention, d’obtenir des spectres de fragmentation MS/MS de référence, de construire des gammes de calibration. De plus des étalons internes isotopiquement marqués, souvent disponibles commercialement.
Spécificités de l’analyse ciblée en protéomique
En protéomique[2], la situation est plus complexe.Les standards de peptides ou de protéines sont souvent difficiles à trouver, coûteux ou inexistants.
Cas où un standard est disponible
Lorsque le peptide ou la protéine pur(e) est disponible sur le marché, l’analyse ciblée peut être mise en œuvre de manière relativement classique, de façon classique comme en métabolomique :
-
détermination des transitions MRM ;
-
optimisation des paramètres instrumentaux ;
-
quantification avec étalon interne si disponible.
Analyse ciblée sans standard
En l’absence de standard, il reste néanmoins possible de détecter un peptide en mode ciblé. D’abord le Q1 est réglé pour filtrer l le m/z de l’ion précurseur le plus abondant (plusieurs essais sont nécessaire pour déterminer le meilleur état de charge). Plusieurs essais sont souvent nécessaires pour déterminer le meilleur état de charge.
Choix des ions fils
La fragmentation des peptides est relativement prévisible, produisant majoritairement des ions b et y. Il existe par ailleurs des simulateurs de fragmentation peptidique, utiles pour anticiper les ions fils les plus probables.
Dans ce contexte, plusieurs ions fils sont sélectionnés afin de :
-
confirmer la présence du peptide ;
-
vérifier que tous les ions fils présentent des pics chromatographiques ;
-
et que ces pics ont le même temps de rétention, critère clé de validation.
Analyse ciblée au niveau protéine intacte
La détection ciblée d’une protéine intacte est nettement plus complexe.,Les principales difficultés sont :
-
l’extraction des protéines à partir d’échantillons biologiques, souvent délicate et à faible rendement ;
-
la détermination de l’état de charge le plus abondant ;
-
le choix des ions fils, une protéine générant un très grand nombre de fragments.
De plus, comme une protéine produit de nombreux ions fils, l’intensité individuelle de chaque ion est faible. Or, en analyse ciblée, seuls les ions fils sélectionnés atteignent le détecteur, ce qui entraîne une forte perte de sensibilité.
Stratégie privilégiée : la protéomique bottom-up
Pour contourner ces limitations, la stratégie la plus utilisée est la protéomique bottom-up.
Principe
La protéine est :
-
digérée enzymatiquement (le plus souvent par la trypsine) ;
-
analysée sous forme de peptides.
Avantages majeurs
-
Les peptides sont plus faciles à extraire que les protéines intactes ;
-
Les spectres MS/MS sont plus simples ;
-
La fragmentation est mieux comprise et plus facile à prédire ;
-
Les transitions MRM sont plus robustes.
Dans ce cadre, la présence d’une protéine peut être confirmée en ciblant plusieurs peptides caractéristiques (en théorie, un seul peptide unique peut suffire à confirmer la présence de la protéine).
Défis liés à la complexité de l’échantillon
Après digestion, le peptide d’intérêt se retrouve dans un milieu extrêmement complexe, contenant des centaines de milliers d’autres peptides.
De plus :
-
le temps de rétention du peptide est souvent inconnu ;
-
deux ou trois transitions MRM peuvent être insuffisantes pour une identification fiable.
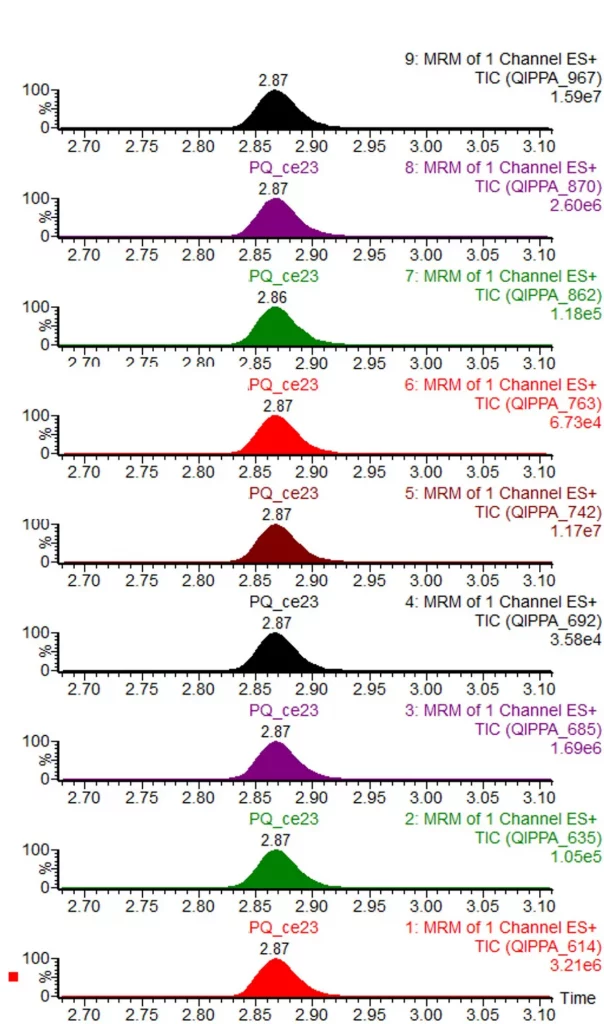
Dans une phase exploratoire, il est donc recommandé :
-
d’augmenter le nombre de transitions surveillées (figure 2) ;
-
afin de confirmer la présence du peptide par la cohérence des pics chromatographiques.
Une fois le peptide identifié et confirmé :
-
les transitions sont optimisées individuellement ;
-
puis seules les deux ou trois meilleures transitions sont conservées pour la méthode finale.
Référence