La spectrométrie de masse en protéomique
La protéomique a pour objectif d’étudier les protéines présentes dans un échantillon biologique, afin de mieux comprendre les mécanismes moléculaires intervenant dans les cellules et les organismes vivants [1]. La spectrométrie de masse (MS) est devenue un outil central dans ce domaine, grâce à sa sensibilité, sa précision et sa capacité à analyser des mélanges complexes.
Les avancées technologiques récentes, notamment le développement de nouveaux analyseurs de masse tels que l’Orbitrap, ainsi que l’introduction de nouvelles méthodes de dissociation comme la dissociation par transfert d’électrons (ETD) [2], ont considérablement élargi les domaines d’application de la protéomique moderne.
L’une des étapes clé en protéomique, est de identifier et quantifier les protéines, pour ce faire deux grandes stratégies analytiques sont utilisées :
-
la stratégie bottom-up, basée sur l’analyse de peptides issus de la digestion enzymatique des protéines,
-
la stratégie top-down, qui repose sur l’analyse directe des protéines intactes.
Bien que la stratégie bottom-up soit la plus répandue en raison de sa robustesse, de son efficacité et de sa simplicité expérimentale, la stratégie top-down permet une caractérisation plus complète des isoformes protéiques et des modifications post-traductionnelles (PTM). Par ailleurs, l’introduction des méthodes de marquage isotopique stable [3] a transformé la spectrométrie de masse d’un simple outil d’identification en un outil quantitatif puissant, capable de mesurer les variations dynamiques d’expression, d’interaction et de modification des protéines.
Stratégie top-down en MS/MS
L’approche top-down consiste à analyser directement un mélange de protéines intactes, sans étape préalable de digestion enzymatique (Figure 1). Les protéines sont d’abord séparées par chromatographie liquide, puis ionisées. Une protéine d’intérêt est ensuite isolée dans le spectromètre de masse, fragmentée dans l’analyseur, et identifiée à partir des valeurs de m/z de l’ion précurseur et de ses ions fragments.
Les principaux avantages de la stratégie top-down sont :
-
la détection des variants de séquence,
-
l’identification des produits de dégradation,
-
la caractérisation précise des combinaisons de modifications post-traductionnelles (PTM).
Cette méthode présente l’avantage de nécessiter moins d’étapes de préparation des échantillons. Elle permet l’analyse de la séquence complète des protéines ainsi que la localisation exacte des modifications post-traductionnelles. De plus, elle offre la possibilité d’obtenir des informations sur les fragments issus de la dégradation partielle des protéines.
Cependant, l’approche top-down comporte plusieurs inconvénients. Elle requiert des spectromètres de masse très performants, à très haut pouvoir de résolution, tels que l’Orbitrap ou le FT-ICR. Le choix des états de charge et des enveloppes isotopiques est complexe, et la fragmentation n’est pas systématique sur l’ensemble des liaisons peptidiques, ce qui peut limiter la couverture de séquence.
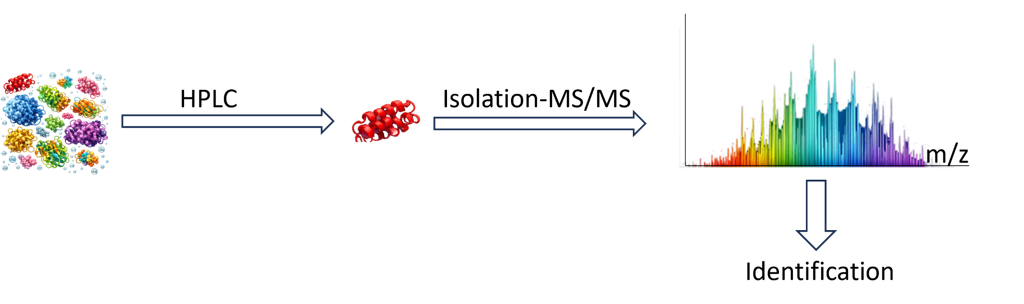
Stratégie bottom-up en MS/MS
La stratégie bottom-up (Figure 2) est actuellement la plus utilisée dans les laboratoires de protéomique. Les protéines sont d’abord digérées par une enzyme protéolytique, généralement la trypsine, afin de générer un mélange de peptides. Ces peptides sont ensuite séparés par chromatographie liquide avant leur analyse par spectrométrie de masse.
Cette séparation préalable permet d’éviter la surcharge du spectromètre de masse et d’améliorer la qualité des spectres acquis. Dans un premier temps, un spectre MS est acquis afin de détecter les peptides présents à un instant donné. Le spectromètre sélectionne ensuite un nombre défini d’ions peptidiques pour les fragmenter successivement : cette approche est appelée TopN, où N correspond au nombre de spectres MS/MS acquis après chaque scan MS.
Le choix de N dépend directement de la vitesse de scan de l’instrument. Un N trop élevé peut conduire à la perte d’informations sur des peptides éluant rapidement, en raison du décalage temporel entre la chromatographie et l’acquisition MS/MS.
Les peptides sont ensuite identifiés à l’aide de logiciels spécialisés (Proteome Discoverer, MaxQuant, etc.). À partir de ces identifications peptidiques, les protéines correspondantes peuvent être inférées. Il est à noter qu’une protéine peut parfois être identifiée sur la base d’un seul peptide unique.
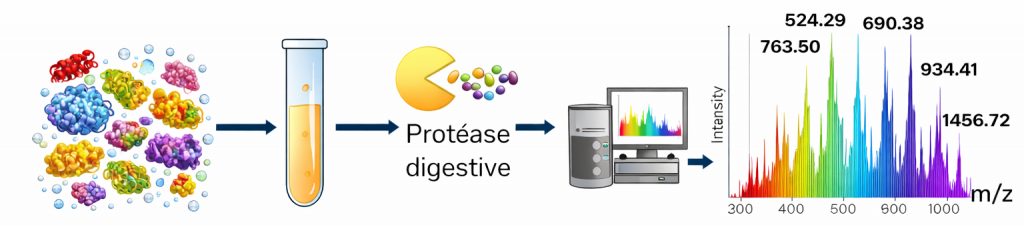
Préparation des échantillons
Les protéines peuvent être digérées directement en solution, puis analysées par LC-MS/MS. Cependant, il est souvent préférable de réaliser une séparation préalable sur gel de polyacrylamide, suivie d’une digestion in-gel.
Cette approche présente plusieurs avantages :
-
purification du mélange protéique,
-
réduction de la complexité de l’échantillon,
-
amélioration de l’identification des peptides.
Après migration électrophorétique, les protéines sont révélées par différentes méthodes (coloration à l’argent, bleu de Coomassie, fluorescence). Les zones d’intérêt sont découpées, puis les protéines qu’elles contiennent sont digérées enzymatiquement. Les peptides obtenus peuvent être conservés plusieurs jours à température ambiante, mais une conservation à long terme nécessite un stockage à basse température.
Analyse LC-MS/MS
La durée d’une analyse LC-MS/MS dépend de la complexité de l’échantillon. Une analyse standard dure environ une heure, mais des gradients chromatographiques plus longs peuvent être utilisés afin d’améliorer la séparation des peptides.
Malgré ces précautions, un échantillon biologique peut contenir des dizaines de milliers de peptides, dont les pics chromatographiques se chevauchent fréquemment. Pour optimiser la sélection des ions à fragmenter, des méthodes dites data-dependent acquisition (DDA) sont utilisées, dans lesquelles les ions sont sélectionnés en fonction de critères tels que leur intensité, leur état de charge ou leur m/z.
Modifications post-traductionnelles
Les modifications post-traductionnelles (PTM) jouent un rôle essentiel dans la régulation des processus cellulaires, la localisation subcellulaire des protéines et la formation de complexes protéiques. Elles constituent l’un des principaux défis de la protéomique moderne.
Les PTM les plus courantes incluent notamment :
-
l’acétylation,
-
la phosphorylation,
-
l’oxydation de la méthionine,
-
la glycosylation,
-
l’ubiquitination.
Certaines modifications nécessitent des stratégies d’acquisition spécifiques. Par exemple, les peptides phosphorylés présentent fréquemment une perte neutre de 98 Da (H₃PO₄) lors de la fragmentation, ce qui peut nécessiter des méthodes de multi-activation pour améliorer leur identification.
Quantification protéomique par marquage isotopique stable
La quantification protéomique par marquage d’isotopes stables repose sur la comparaison de protéines ou de peptides possédant exactement la même séquence, mais différant par leur masse, en raison de l’incorporation d’isotopes lourds stables tels que le 15N ou le 13C. Les protéines ou peptides ainsi comparés présentent des propriétés physicochimiques identiques ; ils sont donc séparés de manière similaire lors de la chromatographie liquide et ionisés avec la même efficacité. Cette approche permet une quantification relative fiable basée sur les intensités des signaux mesurés en spectrométrie de masse.
Il existe plusieurs méthodes de quantification par marquage isotopique, parmi lesquelles les plus utilisées sont : SILAC (Stable Isotope Labeling with Amino acids in Cell culture), iTRAQ (Isobaric Tags for Relative and Absolute Quantification) et TMT (Tandem Mass Tags).
SILAC
La méthode SILAC repose sur la culture de deux populations cellulaires dans des milieux distincts. La première population est cultivée dans un milieu contenant des acides aminés naturels dits « légers », tandis que la seconde est cultivée dans un milieu enrichi en acides aminés marqués avec des isotopes lourds stables (non radioactifs). Par exemple, l’arginine peut être marquée avec du 13C sur ses six atomes de carbone en remplacement du 12C naturel.
Au cours de la croissance cellulaire, les acides aminés lourds sont incorporés dans l’ensemble des protéines synthétisées. Ainsi, les peptides contenant une arginine lourde présentent une différence de masse de 6 Da par rapport à leurs homologues légers. D’autres schémas de marquage utilisant le 13C ou le 15N peuvent également être employés.
L’un des principaux avantages de la méthode SILAC est que les protéines issues des deux populations cellulaires peuvent être mélangées précocement et analysées simultanément par spectrométrie de masse. Les peptides chimiquement et physiquement identiques, mais différant par leur composition isotopique, sont distingués par leur différence de masse. Le rapport des intensités des pics correspondants dans le spectre de masse reflète directement le rapport d’abondance relative des protéines (figure 3).
Cependant, la méthode SILAC présente certaines limitations. Elle nécessite une préparation longue et coûteuse, et le nombre d’échantillons comparables est restreint. De plus, lorsque la différence d’abondance entre deux peptides est très importante, le peptide majoritaire peut provoquer une surcharge de l’analyseur, entraînant un effet de charge d’espace. Dans les analyseurs de type Orbitrap, les ions présents en faible quantité ont un temps de cohérence plus court, ce qui peut conduire à une sous-estimation de leur intensité réelle.
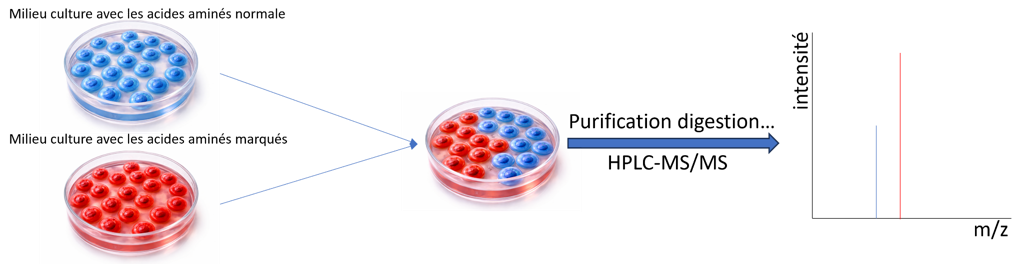
iTRAQ
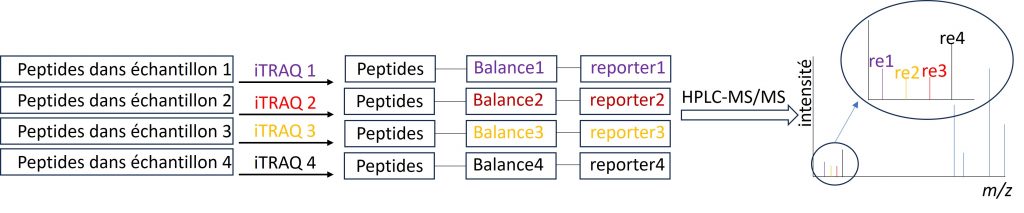
Le marquage isotopique iTRAQ (Isobaric Tags for Relative and Absolute Quantification) permet l’identification et la quantification relative des protéines in vitro à partir de plusieurs échantillons analysés simultanément par spectrométrie de masse en tandem (MS/MS). Contrairement à la méthode SILAC, iTRAQ ne nécessite pas de culture cellulaire dans des milieux spécifiques et permet la comparaison de jusqu’à quatre échantillons (ou davantage selon les versions) au cours d’une seule analyse LC-MS/MS.
Les peptides sont marqués chimiquement sur leurs groupements amines à l’aide de réactifs iTRAQ, qui sont constitués de trois éléments :
- un groupement reporteur, portant une masse spécifique (généralement comprise entre 114 et 117 Da) ;
- une balance de masse neutre, assurant que les peptides marqués restent isobares au niveau MS ;
- un groupement réactif, permettant la fixation covalente du marqueur sur les peptides. (figure 4)
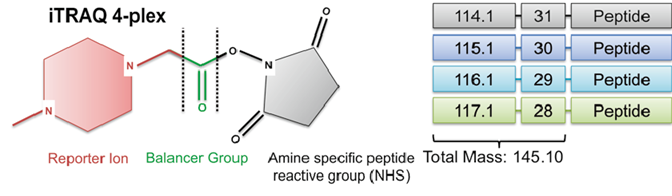
Lors de la fragmentation en MS/MS, les ions reporteurs sont libérés et détectés, permettant une quantification relative basée sur l’intensité de leurs signaux (figure 5).
TMT (Tandem Mass Tag)
Le principe de quantification par TMT repose sur le même mécanisme que celui de iTRAQ. Cependant, les groupements reporter et balance (ou groupements de normalisation) sont de plus grande taille, ce qui permet d’introduire un plus grand nombre de variations de masse. Cette caractéristique rend possible la comparaison simultanée d’un nombre plus élevé d’échantillons, jusqu’à 32 échantillons avec la technologie TMTpro.
Quantification Label free
Bien que ces méthodes ne se limitent pas à la comparaison de deux échantillons, plusieurs échantillons peuvent être analysés simultanément. Toutefois, pour une approche plus économique et applicable à un nombre illimité d’échantillons, la quantification label-free est souvent privilégiée.
La méthode la plus couramment utilisée repose sur la chromatographie des ions extraits (extracted ion chromatography, XIC)¹²˒¹³, qui permet de calculer l’aire sous le pic chromatographique et de comparer ces valeurs entre différents échantillons. L’une des principales limites de cette approche est que les peptides sont analysés lors de différentes acquisitions, ce qui la rend sensible aux variations instrumentales et aux effets de matrice.
Récemment, une méthode de quantification label-free a suscité un intérêt important : le spectral counting. Toutefois, cette approche présente des limites majeures qui remettent en question sa validité pour une quantification fiable. Le principe du spectral counting repose sur le nombre de spectres MS/MS attribués à un même peptide (ou une même protéine), ces nombres étant ensuite comparés entre différents échantillons. Cependant, cette méthode est fondamentalement biaisée pour plusieurs raisons. Tout d’abord, pour qu’un peptide soit sélectionné de manière répétée pour la fragmentation, il doit être présent à une concentration très élevée. De plus, il est nécessaire de désactiver l’exclusion dynamique afin qu’un peptide présentant le signal chromatographique le plus intense soit fragmenté plusieurs fois, au détriment d’autres peptides coéluant. Un problème fondamental de cette approche est qu’elle ne prend jamais en compte l’intensité du pic chromatographique. Or, dans toutes les méthodes de quantification reposant sur la chromatographie couplée à la spectrométrie de masse, cette dimension est essentielle, qu’elle soit utilisée seule (intensité) ou combinée à la largeur du pic pour le calcul de l’aire sous la courbe. Il est méthodologiquement impossible d’ignorer cette information sans compromettre la quantification.
Par ailleurs, le mécanisme expliquant la relation entre le nombre de spectres MS/MS et l’abondance réelle des peptides n’est pas clairement établi. En pratique, une concentration élevée de peptide peut conduire à un temps de remplissage plus court dans des analyseurs de type Orbitrap, ce qui peut augmenter la fréquence des scans MS/MS. Toutefois, le temps de remplissage n’est pas le seul facteur déterminant la durée d’un scan MS/MS complet. À titre d’exemple, un cycle MS/MS comprend à la fois un temps de remplissage de l’ion trap (environ 10 ms) et un temps d’acquisition des ions (environ 20 ms). Même lorsque la concentration du peptide est extrêmement élevée et que le temps de remplissage devient négligeable, le temps d’enregistrement des ions demeure incompressible. Ainsi, le nombre de scans MS/MS ne peut pas être considéré comme un reflet direct et linéaire de l’abondance des peptides.