
La spectrométrie de masse est une technique analytique capable de fournir des informations à la fois qualitatives (structure) et quantitatives (quantité ou concentration). Au cours des dernières décennies, la source d’ionisation électrospray (ESI) s’est imposée comme une technique majeure dans les laboratoires cliniques. Elle constitue un outil sensible, robuste et fiable pour la recherche clinique et permet un couplage efficace avec la chromatographie liquide haute performance (HPLC).
Le couplage HPLC-ESI-MS est devenu une technique très puissante, capable d’analyser des petites comme des grosses molécules, de polarités variées, au sein d’échantillons biologiques complexes. La spectrométrie de masse en tandem (MS/MS) permet de sélectionner un ion précurseur et de le fragmenter afin de produire des ions fragments. L’analyse de ces fragments permet soit d’élucider la structure d’une molécule inconnue, soit de confirmer l’identité d’une molécule cible avec une grande spécificité.
La spectrométrie de masse haute résolution (HRMS) est aujourd’hui de plus en plus accessible, tant en termes de coût que d’utilisation. Elle apporte une dimension supplémentaire à la spectrométrie de masse en permettant la mesure de la masse précise, ce qui améliore considérablement la capacité d’identification et de confirmation des molécules tout en conservant une excellente sensibilité.
En recherche clinique, les spectromètres de type MALDI-TOF MS sont particulièrement répandus, notamment en microbiologie. Ils permettent une identification rapide et fiable des micro-organismes à partir de profils de masses, avec une grande facilité d’utilisation. Par ailleurs, la source plasma à couplage inductif (ICP-MS) est largement utilisée pour l’analyse des éléments inorganiques et des métaux, grâce à son efficacité d’ionisation très élevée, proche de 100 % pour la plupart des éléments.
La puissance de la spectrométrie de masse, associée à la diversité des sources d’ionisation et des analyseurs de masse, permet d’adapter cette technique à une grande variété de molécules. Cette polyvalence fait de la spectrométrie de masse un outil incontournable en recherche clinique, aussi bien pour l’identification, la quantification que la caractérisation structurale des biomolécules.
La chromatographie liquide couplée à la spectrométrie de masse (LC-MS)
Pour l’analyse d’échantillons biologiques complexes tels que le sérum ou le plasma, la chromatographie liquide (LC) est largement utilisée car elle peut être couplée en ligne à la spectrométrie de masse. Ce couplage permet une séparation efficace des constituants avant leur détection et améliore fortement la spécificité et la sensibilité de l’analyse.
Par exemple, la quantification précise de protéines à très faible concentration (de l’ordre du ng/mL) est réalisée de manière reproductible en utilisant la chromatographie liquide en phase inverse (RP-LC) pour séparer les peptides cibles, suivie d’une analyse par spectrométrie de masse en mode MRM (Multiple Reaction Monitoring) ou SRM (Selected Reaction Monitoring) sur des spectromètres de type triple quadripôle [1]. La principale limitation de cette approche est le temps d’analyse, souvent élevé en raison des gradients chromatographiques longs nécessaires à une séparation optimale.
Une alternative pour certaines applications cliniques est le couplage de l’électrophorèse capillaire (CE) à la spectrométrie de masse, qui est particulièrement utilisée pour l’analyse des échantillons urinaires [2,3]. Dans le cadre du diagnostic, une quantification relative de l’excrétion de biomarqueurs est souvent suffisante pour évaluer les profils de polypeptides urinaires avant les étapes de reconnaissance et de classification.
La quantification ciblée des protéines et des peptides peut être réalisée avec différents types de spectromètres de masse, notamment les MALDI-TOF MS [4], les pièges à ions, les quadripôles simples [5], les systèmes Q-TOF, ainsi que les spectromètres de haute résolution tels que les Orbitrap [6,7]. Cependant, dans la pratique clinique et bioanalytique, les triple quadripôles (QQQ) restent les instruments les plus utilisés pour la quantification ciblée, en raison de leur excellente sensibilité, spécificité et robustesse en mode SRM/MRM [8]. Les instruments QQQ modernes permettent de cibler et de quantifier des centaines de peptides simultanément dans le plasma au cours d’une seule analyse [9].
Les performances du couplage LC-MS sont encore améliorées par l’utilisation de l’ultra-haute performance chromatographie liquide (UHPLC) associée aux spectromètres de masse de haute résolution (HRMS). La HRMS fournit des mesures de masse exacte et une très haute résolution, ce qui améliore fortement l’identification et la confirmation des composés. Contrairement aux systèmes QQQ, la HRMS permet des approches non ciblées ou semi-ciblées en plus des analyses MS/MS. Bien que ces instruments offrent une grande polyvalence et des performances analytiques élevées, leur coût d’acquisition et de maintenance reste significativement plus élevé.
MALDI-TOF MS : application en microbiologie de routine
De nombreux laboratoires de microbiologie hospitalière et de biologie médicale se sont récemment équipés de spectromètres MALDI-TOF MS, remplaçant progressivement les méthodes phénotypiques classiques. L’implantation de cette technologie en routine ne pose généralement pas de difficultés majeures.
Les instruments MALDI-TOF MS sont robustes et peu sensibles à la contamination, car ils produisent essentiellement des ions monocationiques, ce qui facilite l’interprétation des spectres. Grâce aux logiciels de pilotage et aux bases de données intégrées, leur utilisation est relativement simple et leur prise en main par le personnel de laboratoire est rapide. Les étapes les plus critiques sur le plan technique concernent la calibration quotidienne, la préparation du mélange échantillon–matrice et son dépôt sur la plaque métallique. Un excès d’échantillon ou un mauvais dépôt peut dégrader la qualité du spectre et compromettre l’identification. Une manipulation soigneuse est également nécessaire pour éviter la contamination croisée entre dépôts adjacents.
Une fois ces paramètres maîtrisés, les performances d’identification dépendent principalement des espèces analysées et de la qualité de la base de données. En routine, les taux d’identification correcte au niveau du genre pour les espèces les plus fréquemment isolées varient de 87 % à 99 % selon les études [10–12]. Les entérobactéries sont particulièrement bien identifiées, avec une identification correcte de l’espèce dans 97 % à 99 % des cas. En revanche, certaines bactéries restent plus difficiles à distinguer, notamment les Shigella spp., dont les profils sont parfois absents ou mal discriminés dans les bases de données, ainsi que les streptocoques du groupe mitis, difficiles à différencier de Streptococcus pneumoniae.
Une autre application importante du MALDI-TOF MS est l’identification directe des bactéries à partir des prélèvements cliniques, notamment des flacons d’hémoculture positifs [13,14] et des urines [15]. Cette approche permet un gain de temps considérable pour la mise en route d’une antibiothérapie adaptée. Pour les hémocultures, l’identification peut être obtenue en une vingtaine de minutes après quelques étapes de préparation visant à éliminer les cellules sanguines. Les taux d’identification correcte au niveau du genre varient alors de 72 % à 98 % selon les études, avec des limitations similaires à celles observées à partir de colonies isolées. En présence d’hémocultures polymicrobiennes, les spectres sont plus complexes et, le plus souvent, seul le germe prédominant peut être identifié.
Pour les urines, les résultats sont très encourageants lorsque la charge bactérienne est suffisante, généralement supérieure à 10⁵ CFU/mL.
Ainsi, le MALDI-TOF MS s’est imposé rapidement et durablement dans les laboratoires de microbiologie clinique. Ses principaux atouts sont sa robustesse, sa rapidité, sa précision d’identification, sa facilité d’utilisation et son faible coût par échantillon, ce qui en fait aujourd’hui une technologie de référence pour l’identification microbienne de routine.
L’imagerie par spectrométrie de masse
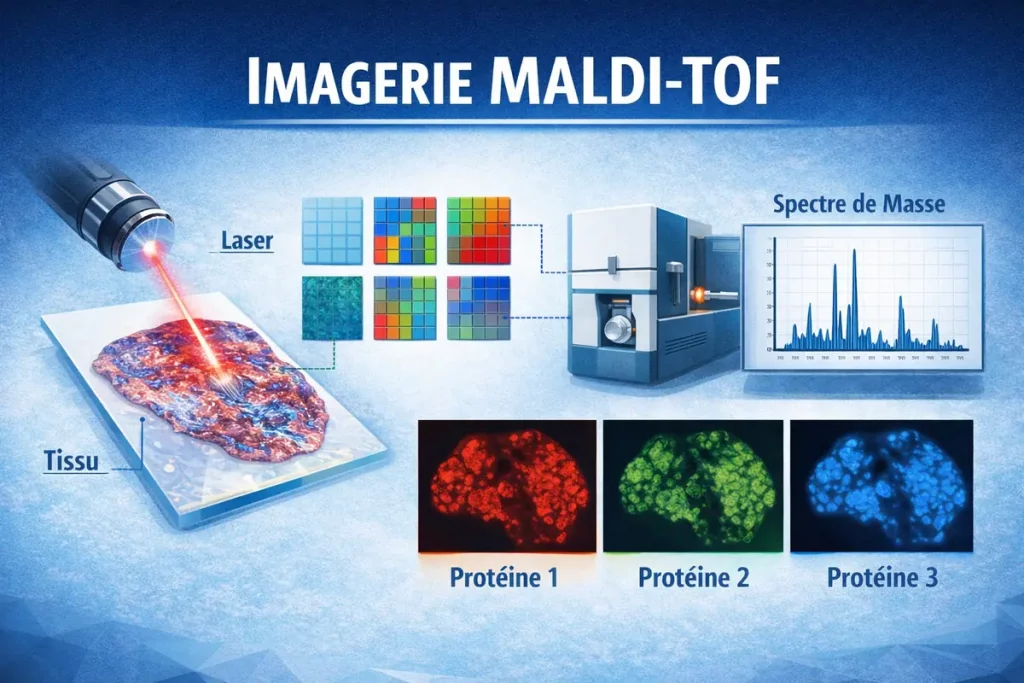 L’imagerie par spectrométrie de masse (MSI) permet d’analyser directement des coupes de tissus et de représenter la distribution spatiale de molécules sélectionnées sans marquage préalable. Deux principales sources d’ionisation sont utilisées pour ces expériences : la MALDI (désorption/ionisation laser assistée par matrice) et la SIMS (Secondary Ion Mass Spectrometry).
L’imagerie par spectrométrie de masse (MSI) permet d’analyser directement des coupes de tissus et de représenter la distribution spatiale de molécules sélectionnées sans marquage préalable. Deux principales sources d’ionisation sont utilisées pour ces expériences : la MALDI (désorption/ionisation laser assistée par matrice) et la SIMS (Secondary Ion Mass Spectrometry).
La MALDI est aujourd’hui la source la plus utilisée en imagerie par spectrométrie de masse en raison de sa polyvalence et de sa capacité à analyser des biomolécules de masses élevées. La procédure commence par la préparation de coupes de tissu d’environ 10 à 20 µm d’épaisseur, réalisées à basse température (environ −15 °C à −20 °C) à l’aide d’un cryostat. Les coupes sont ensuite déposées sur des lames conductrices, généralement en verre recouvert d’une fine couche d’oxyde d’indium-étain (ITO) ou sur des plaques en acier inoxydable. Avant l’analyse, les échantillons sont ramenés à température ambiante et séchés sous atmosphère contrôlée.
L’étape la plus critique de l’imagerie MALDI est le dépôt de la matrice, qui conditionne à la fois la sensibilité, la reproductibilité et la résolution spatiale. Plusieurs méthodes sont utilisées : nébulisation, dépôt automatisé de microgouttelettes par robot, ou dépôt manuel à la pipette de matrices spécifiques. La nébulisation permet d’obtenir un dépôt relativement homogène avec des cristaux de quelques micromètres, mais nécessite un compromis entre la volatilité du solvant et le temps de séchage afin de limiter la délocalisation des analytes.
Les matrices les plus couramment utilisées sont l’acide α-cyano-4-hydroxycinnamique (CHCA) pour les petites molécules et les peptides de faible masse (généralement < 3000 Da), et l’acide sinapinique pour les peptides et protéines de plus grande taille. Les systèmes automatisés déposent des gouttelettes typiquement espacées de 150 à 200 µm, avec un diamètre minimal d’environ 100–150 µm, ce qui offre une bonne sensibilité mais limite la résolution spatiale. Plus récemment, l’utilisation de matrices ioniques solides (par exemple des mélanges équimolaires de CHCA et d’aniline) a simplifié la préparation des échantillons, permettant un dépôt direct à la pipette tout en améliorant la sensibilité et l’homogénéité.
La résolution latérale en imagerie MALDI est principalement déterminée par la taille du spot laser, typiquement comprise entre 20 et 50 µm sur les instruments commerciaux, ce qui est supérieur à la taille d’une cellule unique. Ainsi, la résolution spatiale est généralement limitée à quelques dizaines de micromètres. Pour chaque pixel, plusieurs tirs laser sont nécessaires afin d’obtenir un spectre de masse représentatif.
Les spectromètres MALDI utilisés en imagerie sont équipés soit de lasers N₂ (337 nm), soit de lasers Nd:YAG triplés (355 nm), fonctionnant respectivement à des fréquences typiques d’environ 20 Hz et jusqu’à 200 Hz ou plus. Par conséquent, l’acquisition d’une image de 100 × 100 pixels (soit 10 000 spectres) peut nécessiter plusieurs heures, typiquement 10 à 20 heures selon la fréquence du laser, le nombre de tirs par pixel et les paramètres d’acquisition.
Plusieurs études portant sur le cancer et les maladies neurodégénératives ont montré que l’imagerie MALDI-MS constitue une technique clé pour l’identification de biomarqueurs, l’analyse de leur localisation spatiale et leur validation [17–19].
L’exploitation des tissus archivés, notamment les échantillons fixés au formol et inclus en paraffine (FFPE) conservés dans les services d’anatomopathologie hospitaliers, représente une véritable mine d’or en termes de données cliniques et biologiques [20].
L’application de l’imagerie MALDI-MS à ces matériaux archivés pourrait permettre la constitution de bases de données internationales de biomarqueurs de maladies, facilitant le diagnostic précoce de nombreuses pathologies ainsi que le suivi de leur progression et de la réponse aux traitements.
La HPLC-ICP-MS pour l’étude des oligo-éléments
La nécessité de déterminer les espèces chimiques (état d’oxydation et forme moléculaire) des oligo-éléments présents dans l’environnement et dans les matrices biologiques a fortement augmenté au cours des dernières décennies, en raison de leur accumulation liée aux activités humaines [21]. Si certains oligo-éléments sont essentiels à la santé humaine, ils peuvent devenir toxiques à des concentrations élevées ou sous certaines formes chimiques [22]. Depuis les premières études menées dans les années 1960 [23], il est désormais bien établi que la toxicité dépend fortement de l’état d’oxydation et de la spéciation chimique, ce qui a conduit au développement de nombreuses recherches en biologie, environnement, pharmacie et en applications cliniques [24,25].
Pour l’analyse de ces éléments, la spectrométrie de masse couplée à une source à plasma inductif (ICP-MS) est la technique de référence. Cette source est particulièrement adaptée à la détection des éléments inorganiques. Le principe de l’ICP repose sur la génération d’un plasma à très haute température (environ 6 000 à 10 000 K), dans lequel les composés sont atomisés et ionisés presque complètement. L’ICP-MS est compatible avec les techniques de séparation en phase liquide, notamment la chromatographie liquide (LC) et la chromatographie liquide haute performance (HPLC), ce qui permet l’analyse d’échantillons complexes et la spéciation des oligo-éléments.
En recherche clinique, trois éléments ont fait l’objet d’une attention particulière au cours des dernières décennies : l’arsenic (As), le sélénium (Se) et le mercure (Hg). Le sélénium est un oligo-élément essentiel, impliqué dans de nombreux processus métaboliques et dans la protection contre le stress oxydatif. En revanche, l’arsenic et le mercure ne sont pas essentiels et sont connus pour leur toxicité, ce qui a conduit à de nombreuses études sur leur présence dans les aliments, l’eau et l’environnement.
De nombreuses investigations basées sur la HPLC-ICP-MS ont permis de mieux comprendre la distribution, la biodisponibilité et la toxicité de ces éléments en fonction de leur spéciation chimique. Delafiori et ses collaborateurs ont notamment synthétisé ces travaux dans une revue détaillant la technique ICP-MS, les mécanismes de toxicité, la localisation biologique des espèces d’As, de Se et de Hg, ainsi que les méthodes d’extraction et de préparation des échantillons.
Conclusion
À l’heure actuelle, les spectromètres de masse triple quadripôle (QQQ) constituent les instruments les plus largement utilisés pour l’analyse LC-MS dans les laboratoires cliniques. Les progrès technologiques de ces systèmes permettent aujourd’hui de surveiller des centaines de transitions MRM au cours d’une seule analyse LC-MS/MS, ce qui en fait des outils particulièrement performants pour la quantification ciblée en recherche clinique. Toutefois, cette approche présente une limitation majeure : elle ne permet pas la détection d’analytes non ciblés, ce qui restreint son utilisation pour le criblage global. Dans ce contexte, les approches basées sur la spectrométrie de masse haute résolution (HRMS) sont mieux adaptées aux stratégies de dépistage et d’analyse non ciblée.
Le triple quadripôle demeure aujourd’hui la référence en matière de précision et de robustesse pour la quantification. Néanmoins, la nouvelle génération d’analyseurs temps de vol (TOF) dédiés à la quantification, tels que la technologie QuanTof™ de Waters, marque une avancée importante dans ce domaine. Par ailleurs, les instruments de type Orbitrap occupent une place croissante dans les laboratoires cliniques, en raison de leur haute résolution, de leur masse exacte et de leur polyvalence analytique.
La HRMS est désormais suffisamment mature pour être intégrée aux laboratoires cliniques pour des applications de criblage, d’identification et de confirmation, notamment grâce à la baisse des coûts d’acquisition et de maintenance ainsi qu’à l’amélioration de l’ergonomie et de la fiabilité des instruments. Il ne fait aucun doute que ces technologies joueront également un rôle de plus en plus important dans la quantification dans un avenir proche, en complément ou en alternative aux systèmes triple quadripôle.
Référence
[8] Pan, S., Aebersold, R., Chen, R., Rush, J., et al., Mass spectrometry based targeted protein quantification: methods and applications. J. Proteome Res. 2009, 8, 787–797.
[9] Burgess, M. W., Keshishian, H., Mani, D. R., Gillette, M. A., Carr, S. A., Simplified and efficient quantification of low-abundance proteins at very highmultiplex via targeted mass spectrometry. Mol. Cell. Proteomics 2014, 13, 1137–1149.
[10] Blondiaux N, Gaillot O, Courcol RJ. MALDI-TOF mass spectrometry to identify clinical bacterial isolates : Evaluation in a teaching hospital. Pathol Biol (Paris) 2010 ; 58 : 55-7.
[11] Van Veen SQ, Claas EC, Kuijper EJ. High-throughput identification of bacteria and yeast by matrixassisted laser desorption ionization mass spectrometry (MALDI-TOF MS) in routine medical microbiology laboratory. J Clin Microbiol 2010 ; 48 : 900-7.
[12] Seng P, Drancourt M, Gouriet F, et al. Ongoing revolution in bacteriology : routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin Infect Dis 2009 ; 49 : 543-51.
[13] Ferroni A, Suarez S, Beretti JL, et al. Real-time identification of bacteria and Candida species in positive blood culture broths by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol 2010 ; 48 : 1542-8.
[14] La Scola B, Raoult D. Direct identification of bacteria in positive blood culture bottles by matrix-assisted laser desorption ionisation time-of-flight mass spectrometry. PLoS One 2009 ; 4 : e8041.
[15] Ferreira L, Sanchez-Juanes F, Gonzalez-Avila M, et al. Direct identification of urinary tract pathogens from urine samples by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol 2010 ; 48 : 2110-5.
[16] Mass Spectrometry Methodology in Lipid Analysis Lin Li, Juanjuan Han, Zhenpeng Wang, Jian’an Liu, Jinchao Wei, Shaoxiang Xiong and Zhenwen Zhao
[17] Dreisewerd, K., Lemaire, R., Pohlentz, G., Salzet, M., Wisztorski, M.,Berkenkamp, S., and Fournier, I. (2007) Molecular profiling of native and MALDI Imaging Mass Spectrometry Molecular & Cellular Proteomics 8.9 2031 matrix-coated tissue slices from rat brain by infrared and ultraviolet laser desorption/ionization orthogonal time-of-flight mass spectrometry. Anal. Chem. 79, 2463–2471
[18] Brown, L. M., Helmke, S. M., Hunsucker, S. W., Netea-Maier, R. T., Chiang, S. A., Heinz, D. E., Shroyer, K. R., Duncan, M. W., and Haugen, B. R. (2006) Quantitative and qualitative differences in protein expression between papillary thyroid carcinoma and normal thyroid tissue. Mol. Carcinog. 45, 613–626
[19] Chaurand, P., Sanders, M. E., Jensen, R. A., and Caprioli, R. M. (2004) Proteomics in diagnostic pathology: profiling and imaging proteins directly in tissue sections. Am. J. Pathol. 165, 1057–1068
[20] Lemaire, R., Desmons, A., Tabet, J. C., Day, R., Salzet, M., and Fournier, I. (2007) Direct analysis and MALDI imaging of formalin-fixed, paraffinembedded tissue sections. J. Proteome Res. 6, 1295–1305
[21] B. Michalke, Element speciationdefinitions, analytical methodology, and some examples, Ecotoxicol. Environ. Saf.56(2003)122–139.
[22] P.J.Parsons, F.Barbosa, Atomic spectrometry and trends in clinical laboratory medicine, Spectrochim. ActaPartB:At.Spectrosc.62(2007)992–1003.
[23] R. Cornelis, J.Caruso, H.Crews, H.Klaus, Handbook of Elemental Speciation: Techniques and Methodology, JohnWiley & Sons,Chichester, England, 2003.
[24] V.L. Dressler,F.G.Antes,C.M.Moreira, D.Pozebon, F.A.Duarte, As, Hg, I, Sb, Se and Sn speciation in body fluids and biological tissues using hyphenated-ICP-MS techniques: Areview, Int. J.MassSpectrom. 307(2011) 149–167.
[25] B. Meermann,M.Sperling, Hyphenated techniques as tools for speciation Analysis of metal-based pharmaceuticals: developments and applications, Anal. Bioanal.Chem.403(2012)1501–1522.
[26] S. Kokarnig, A.Tsirigotaki, T.Wiesenhofer, V.Lackner, K.A.Francesconi,S. A.Pergantis, D.Kuehnelt, Concurrent quantitative HPLC-mass spectrometry profiling of small selenium specie s in human serum and urine after ingestion of selenium supplements, J.TraceElementsMed.Biol.29(2015)83–90.
[27] M. Molin, S.M.Ulven, L.Dahl, V.H.Telle-Hansen, M.Holck, G.Skjegstad, O. Ledsaak, J.J.Sloth, W.Goessler, A.Oshaug, J.Alexander, D.Fliegel,T. A.Ydersbond, H.M.Meltzer, Humans seem to produce arsen obetaine and Dimethylarsinateafterabolus dose of seafood, Environ.Res.112(2012) 28–39.
[28] M. Vinceti,P.Grill, C.Malagoli,T.Filippini,S.Storani,M.Malavolti, B. Michalke, Selenium speciation in human serum and it simplications for epidemiologic research:across-sectionalstudy, J.TraceElementsMed.Biol. 31(2015)1–10.
[29] M. Contreras-Acuña,T.García-Barrera,M.A.García-Sevillano,J.L.Gómez- Ariza, Arsenic metabolites in human serum and urine after seafood (Ane-monia sulcata) consumption and bio accessibility assessment using liquid chromatography coupled to inorganic and organic mass spectrometry, Mi-crochem. J.112(2014)56–64
[30] M. Molin,S.M.Ulven,L.Dahl,W.Goessler, D.Fliegel, M.Holck, J.J.Sloth, A.Oshaug, J.Alexander, H.M.Meltzer, T.A.Ydersbond, Urinary excretion of arsenicals following daily intake of various seafoods during at wo weeks intervention,FoodChem. .66(2014)76–88.
[31] B. Chen,F.Cao,C.Yuan,X.Lu, S.Shen,J.Zhou, X.C.Le, Arsenic speciationin Saliva of acute promyelocytic leukemia patients undergoing arsenic trioxide treatment, Anal. Bioanal. Chem. 405(2013)1903–1911.
[32] S.S. deSouza, A.D.Campiglia, F.BarbosaJr.,Asimplemethodformethyl- mercury, inorganic mercury and ethylmercury determination in plasma samples by high performance liquid chromatography- cold-vapor-inductively coupled plasma mass spectrometry, Anal. Chim.Acta761(2013)11–17.
[33] F.Moreno,T.Garcia-Barrera,J.L.Gomez-Ariza,Simultaneousspeciation and Preconcentration of ultra trace concentrations of mercury and selenium species inenvironmental and biological samples by hollow fiber liquid phase Micro extraction prior to high performance liquid chromatography coupled to inductively coupled plasma mass spectrometry, J.Chromatogr.A1300 (2013)43–50.
[34] J. Scheer,S.Findenig,W.Goessler, K.A.Francesconi, B.Howard, J.G.Umans, J. Pollak, M.Tellez-Plaza, E.K.Silbergeld, E.Guallar, A.Navas-Acien, Arsenic species and selected metals in human urine: validation of HPLC/ICPMS and ICPMS procedures for along-term population-based epidemi ological study, Anal. Methods4 (2012)406–413.