Application de la spectrométrie de masse dans le domaine de l’antidopage
 À l’origine, la lutte contre le dopage était principalement assurée par le Comité international olympique (CIO). En 1999, l’Agence mondiale antidopage (World Anti-Doping Agency, WADA) a été créée afin d’harmoniser et de renforcer les contrôles à l’échelle mondiale. Cet organisme est aujourd’hui reconnu par l’ensemble des fédérations sportives internationales. On compte actuellement 34 laboratoires accrédités par la WADA dans le monde, habilités à effectuer les analyses d’échantillons de contrôle du dopage, et de nombreux autres laboratoires sont engagés dans une procédure d’accréditation.
À l’origine, la lutte contre le dopage était principalement assurée par le Comité international olympique (CIO). En 1999, l’Agence mondiale antidopage (World Anti-Doping Agency, WADA) a été créée afin d’harmoniser et de renforcer les contrôles à l’échelle mondiale. Cet organisme est aujourd’hui reconnu par l’ensemble des fédérations sportives internationales. On compte actuellement 34 laboratoires accrédités par la WADA dans le monde, habilités à effectuer les analyses d’échantillons de contrôle du dopage, et de nombreux autres laboratoires sont engagés dans une procédure d’accréditation.
L’une des principales difficultés du contrôle antidopage réside dans l’identification de composés interdits au sein de matrices biologiques complexes, alors que ces substances sont souvent présentes à des concentrations extrêmement faibles, parfois proches des limites de détection. Avant l’introduction de la spectrométrie de masse, peu de techniques analytiques offraient à la fois la sensibilité, la spécificité et la fiabilité nécessaires. À cette époque, le contrôle des agents dopants était donc relativement limité, d’autant plus que la liste des substances interdites était courte et que les pratiques de dopage restaient peu sophistiquées.
Au cours des dernières décennies, les progrès technologiques ont été spectaculaires, mais le dopage a évolué en parallèle : en une quarantaine d’années, la liste des substances interdites a été multipliée par environ dix. Jusqu’aux années 1970, le dopage reposait essentiellement sur des composés exogènes (par exemple la morphine, les amphétamines ou les stéroïdes synthétiques), de masse moléculaire comprise typiquement entre 200 et 500 Da, qui ne sont pas produits naturellement par l’organisme et sont relativement faciles à détecter. Aujourd’hui, de nombreux agents dopants sont des molécules endogènes telles que la testostérone, l’hormone de croissance ou l’érythropoïétine, ce qui complique considérablement leur détection, notamment la distinction entre production physiologique et administration exogène.
Depuis les Jeux Olympiques de Munich en 1972, la spectrométrie de masse est devenue un outil central dans la lutte contre le dopage [1]. Le couplage de la chromatographie en phase gazeuse à la spectrométrie de masse (GC-MS) s’est rapidement imposé comme une technique de référence. Aujourd’hui, la spectrométrie de masse est indispensable au contrôle antidopage, tant pour l’efficacité analytique que pour la sécurité juridique des athlètes. Elle s’est enrichie de multiples approches, notamment la spectrométrie de masse en tandem (MS/MS), la spectrométrie de masse haute résolution (HRMS) et la spectrométrie de masse à rapports isotopiques (IRMS), utilisée pour distinguer les hormones endogènes des formes synthétiques.
Le développement, à partir des années 1980, des sources d’ionisation à pression atmosphérique (en particulier l’électrospray) a permis le couplage de la chromatographie liquide à la spectrométrie de masse (LC-MS), ouvrant la voie à l’analyse de composés non volatils et thermolabiles, ainsi qu’à des molécules de plus grande taille, y compris des peptides et des protéines. Depuis lors, les méthodes basées sur la spectrométrie de masse n’ont cessé d’évoluer afin d’offrir une sensibilité, une sélectivité et une capacité de criblage toujours accrues, indispensables pour détecter un nombre croissant d’agents dopants présents à des concentrations de plus en plus faibles.
Protocole de collecte et d’analyse des échantillons
Chaque échantillon biologique recueilli lors d’un contrôle antidopage est divisé en deux portions, appelées communément échantillon A et échantillon B.
L’échantillon A est utilisé pour les analyses de criblage (dépistage) et, le cas échéant, pour les analyses de confirmation. Si une substance ou une méthode interdite est détectée, l’échantillon B peut être analysé dans le cadre d’une contre-expertise, à la demande de l’athlète.
L’analyse de l’échantillon A vise à détecter la présence de substances interdites, de leurs métabolites, ou de marqueurs indiquant l’usage d’une substance ou d’une méthode prohibée. La phase de criblage doit permettre la détection du plus grand nombre possible de composés dans une matrice biologique complexe (urine, plasma, sérum). Cette étape doit être rapide, sensible et suffisamment sélective, afin de permettre une réponse dans des délais compatibles avec le calendrier des compétitions, où les intervalles entre épreuves peuvent être très courts.
Afin d’éviter les faux positifs, toute détection suspecte lors du criblage doit être suivie d’une procédure de confirmation. Les analyses de confirmation sont réalisées à partir d’une nouvelle aliquote de l’échantillon A et doivent être au moins aussi sélectives que l’analyse initiale. Elles fournissent des données analytiques complémentaires (par exemple MS/MS, HRMS ou IRMS) permettant d’établir de manière formelle la présence d’une substance interdite ou d’un marqueur de dopage.
La préparation des échantillons
La procédure de préparation des échantillons en analyse antidopage est similaire à celle utilisée en métabolomique, car la recherche de substances interdites repose le plus souvent sur la détection des composés parents et surtout de leurs métabolites.
Dans certains cas, une simple dilution de l’échantillon peut être suffisante pour permettre la détection d’un large éventail de composés. Cependant, les substances dopantes et leurs métabolites sont généralement présents à des concentrations très faibles, souvent de l’ordre du ng/mL ou inférieur, ce qui rend nécessaire une étape d’extraction et de préconcentration afin d’améliorer la sensibilité et la fiabilité de l’analyse.
Des techniques telles que l’extraction en phase solide (SPE), l’extraction liquide–liquide (LLE) ou des méthodes hybrides sont ainsi couramment utilisées pour isoler, nettoyer et concentrer les analytes d’intérêt avant leur analyse par GC-MS/MS, LC-MS/MS ou HRMS.
Injection diluée
Grâce aux progrès technologiques récents, les méthodes de séparation chromatographique sont devenues plus performantes et les spectromètres de masse plus sensibles, rapides et robustes. Dans ce contexte, la simple dilution de l’échantillon urinaire avant injection en LC-MS est de plus en plus utilisée pour le dépistage des agents dopants [2].
Les échantillons d’urine sont simplement dilués avec de l’eau ultrapure ou une solution tampon appropriée, puis directement injectés dans le système LC-MS. L’automatisation de l’injection permet le traitement d’un grand nombre d’échantillons en routine. Cette approche est particulièrement adaptée aux analyses rapides et multi-analytes, car elle nécessite un temps minimal de préparation et permet un débit analytique élevé [3,4].
De plus, la dilution réduit les effets de matrice et permet souvent une meilleure reproductibilité des temps de rétention par rapport aux méthodes impliquant des étapes d’extraction et de préconcentration.
En revanche, cette approche conduit à la détection d’un grand nombre de composés, notamment issus de la matrice urinaire, ce qui peut complexifier l’interprétation des résultats. Par ailleurs, certaines substances présentes à très faible concentration ou fortement affectées par les effets de matrice peuvent être partiellement masquées ou ne pas être détectées sans étape de préconcentration.
Le couplage de la chromatographie en phase gazeuse et de la spectrométrie de masse (GC-MS)
Le couplage de la chromatographie en phase gazeuse (GC) à la spectrométrie de masse (MS) est une technique analytique utilisée depuis plusieurs décennies et largement répandue dans les domaines du contrôle antidopage, de la toxicologie et de l’analyse environnementale. Pour être analysé par GC, l’échantillon doit être volatilisé ou rendu volatil après dérivatisation. Les composés sont ensuite entraînés par un gaz vecteur — le plus souvent l’hélium (et non l’azote en routine moderne) — à travers une colonne capillaire, où ils sont séparés en fonction de leurs propriétés physico-chimiques (volatilité, polarité, interactions avec la phase stationnaire).
La source d’ionisation la plus couramment utilisée en GC-MS est l’ionisation électronique (EI). Cette source exige que les molécules soient thermostables et volatiles, mais elle présente un avantage majeur : elle est hautement reproductible et standardisée. L’ionisation par EI est très énergétique (70 eV), ce qui provoque une fragmentation extensive des molécules. Ainsi, chaque composé produit un spectre de masse caractéristique, composé d’un ion moléculaire (lorsqu’il est stable) et d’un ensemble d’ions fragments spécifiques.
L’un des grands atouts de l’ionisation EI est que, pour une molécule donnée, le spectre obtenu est pratiquement identique quel que soit l’instrument utilisé. Cette propriété permet une identification extrêmement fiable par comparaison avec de grandes bibliothèques de spectres de référence (telles que NIST ou Wiley), ce qui fait du GC-MS une technique de choix pour l’analyse et la confirmation des substances dopantes, des drogues et de nombreux composés organiques volatils.
Le couplage de la chromatographie liquide et de la spectrométrie de masse (LC-MS)
Les techniques d’ionisation à pression atmosphérique (API) ont rendu possible le couplage efficace de la chromatographie en phase liquide (LC) à la spectrométrie de masse (MS). La LC-MS est aujourd’hui l’une des techniques les plus utilisées en bioanalyse et en antidopage, car elle permet l’analyse de composés non volatils, thermolabiles ou de masse moléculaire élevée, sans nécessiter d’étape préalable d’hydrolyse ou de dérivatisation.
La chromatographie liquide offre une sélectivité très élevée grâce à la diversité des phases stationnaires et des phases mobiles disponibles, ainsi qu’à la variété des mécanismes d’interaction possibles (hydrophobes, polaires, ioniques, etc.). Cette flexibilité confère à la LC un avantage majeur par rapport à la chromatographie en phase gazeuse pour l’analyse de molécules polaires ou de grande taille.
Dans les premières applications antidopage, la LC était souvent couplée à des détecteurs UV, mais cette approche ne présentait pas une sélectivité et une sensibilité suffisantes par rapport à la GC-MS [5]. Le développement des sources d’ionisation API a permis de coupler efficacement la LC à la MS et d’ouvrir la voie à des méthodes beaucoup plus performantes pour l’analyse des agents dopants.
Les principales sources API sont l’électrospray (ESI), l’ionisation chimique à pression atmosphérique (APCI) et la photoionisation à pression atmosphérique (APPI). Ces sources présentent des domaines d’application complémentaires. L’APCI est particulièrement adaptée à l’ionisation de composés peu polaires et relativement volatils, tandis que l’APPI, bien que moins utilisée en bioanalyse, constitue une alternative intéressante pour certains analytes hydrophobes. Ces techniques sont dites douces, car elles génèrent peu de fragmentation, produisent des ions moléculaires stables et permettent de limiter les effets de matrice.
L’ESI est la source la plus largement utilisée en LC-MS, car elle permet l’ionisation d’analytes sur une très large gamme de masses, en modes positif et négatif, avec une excellente sensibilité. Son principe repose sur l’application d’une différence de potentiel élevée (typiquement 2 à 6 kV) entre la sortie de la colonne LC et l’entrée du spectromètre de masse, ce qui génère un aérosol de gouttelettes chargées. Un gaz de nébulisation peut être ajouté pour faciliter la formation du spray.
Sous l’effet combiné de la chaleur et de la répulsion coulombienne, les gouttelettes se fragmentent progressivement par évaporation et fissions successives jusqu’à la formation d’ions en phase gazeuse, issus des analytes, parfois associés à quelques molécules de solvant. Ces ions sont ensuite transférés vers l’analyseur de masse pour leur détection et leur identification.
Le couplage de l’électrophorèse capillaire (CE) et l’analyse des protéines
Pour analyser des échantillons biologiques contenant des protéines, il est essentiel de les séparer de la matrice (sang, plasma, sérum, urine) afin de limiter les interférences et de protéger les systèmes analytiques. La précipitation des protéines est une méthode simple et rapide couramment utilisée pour cette étape de préparation. Elle consiste généralement à ajouter un solvant organique (le plus souvent l’acétonitrile ou le méthanol) à l’échantillon biologique, ce qui provoque la dénaturation et la précipitation des protéines. Après centrifugation, le surnageant contient les petites molécules d’intérêt, tandis que les protéines sont éliminées sous forme de culot.
L’élimination des protéines est indispensable avant une analyse par GC-MS ou LC-MS/MS, afin d’éviter l’encrassement des colonnes chromatographiques, la contamination des sources d’ionisation et la perte de sensibilité. Pour le traitement de grands nombres d’échantillons, des plaques de précipitation de protéines sont disponibles. Elles permettent d’automatiser la préparation et d’éviter les transferts manuels, puisque la filtration peut être réalisée directement dans le même puits que l’étape de précipitation [7].
En revanche, lorsque l’objectif est de détecter et caractériser directement des protéines ou des peptides, l’électrophorèse capillaire (CE) constitue une méthode de séparation particulièrement performante, grâce à sa très haute efficacité, sa faible consommation d’échantillon et sa compatibilité avec la spectrométrie de masse. Le couplage CE-MS permet ainsi l’analyse de biomacromolécules avec une grande résolution.
Dans le domaine de l’antidopage, le CE-MS a joué un rôle important, notamment lors des Jeux Olympiques d’Athènes en 2004, où il a été utilisé à grande échelle pour la détection de l’hormone de croissance recombinante, administrée illégalement chez certains athlètes. Cette approche reposait sur la séparation et l’identification de formes protéiques spécifiques, impossibles à distinguer par les seules techniques chromatographiques classiques.
Le dépistage de la testostérone
La testostérone (T) est une hormone sexuelle stéroïdienne synthétisée par l’organisme à partir du cholestérol. Elle est métabolisée puis éliminée dans l’urine sous différentes formes, notamment en présence de son épimère, l’épitestostérone (E) (figure 1). Chez un individu non dopé, le rapport testostérone/épitestostérone (T/E) reste relativement constant. En revanche, lors d’une administration exogène de testostérone, ce rapport augmente de manière significative.
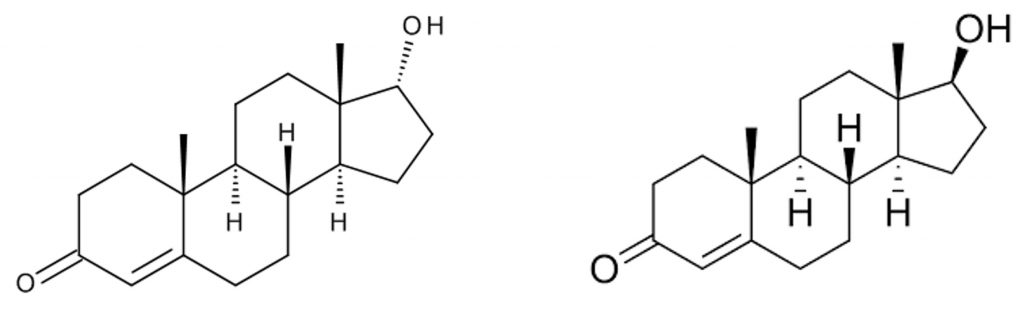
Historiquement, le Comité International Olympique (CIO), puis l’Agence Mondiale Antidopage (WADA), ont retenu le rapport T/E comme principal critère de dépistage du dopage à la testostérone. Toutefois, dans certains cas, ce rapport peut être influencé par des variations physiologiques, génétiques ou pathologiques, ce qui rend la preuve du dopage difficile sur la seule base de ce paramètre.
Les contrôles antidopage visant la testostérone reposent sur une approche en deux étapes :
-
une phase de criblage,
-
une phase de confirmation.
Lors du criblage, la prise de testostérone est suspectée à partir du rapport T/E, mesuré le plus souvent par GC-MS après extraction et dérivatisation des stéroïdes urinaires. Le rapport est calculé à partir des aires ou des hauteurs de pics chromatographiques correspondant à la testostérone et à l’épitestostérone.
Rapport testostérone / épitestostérone (T/E)
La testostérone et l’épitestostérone appartiennent à une classe particulière d’isomères appelés stéréoisomères, et plus précisément des épimères. Deux molécules sont dites isomères lorsqu’elles possèdent la même formule brute mais une organisation spatiale différente. Les épimères sont des stéréoisomères qui ne diffèrent que par la configuration d’un seul carbone asymétrique.
L’épitestostérone est l’épimère C17 de la testostérone : ces deux molécules ne diffèrent que par l’orientation du groupe hydroxyle (–OH) porté par le carbone 17. Elles ont donc la même masse moléculaire, mais leurs propriétés physico-chimiques diffèrent légèrement. Cette différence suffit à leur conférer des temps de rétention distincts en chromatographie gazeuse, ce qui permet leur séparation et leur quantification individuelle par GC-MS.
Cette séparation chromatographique est la base de la mesure fiable du rapport T/E, utilisé comme indicateur de la possible administration exogène de testostérone.
Limites du rapport T/E et critères de positivité
Le critère de positivité du dépistage de la testostérone est défini par l’Agence Mondiale Antidopage (AMA/WADA). Historiquement, un rapport testostérone/épitestostérone (T/E) supérieur à 6 était considéré comme suspect d’une administration exogène de testostérone. Ce seuil a ensuite été abaissé à 4, valeur actuellement utilisée pour déclencher des investigations complémentaires.
Cependant, la fiabilité du rapport T/E est sujette à controverse. En effet, ce rapport présente une variabilité interindividuelle importante, liée notamment à des facteurs génétiques, ethniques et physiologiques. Par exemple, des valeurs moyennes d’environ 0,5 sont observées chez certaines populations asiatiques, alors que des valeurs proches de 1,2 à 1,3 sont plus fréquentes chez les populations caucasiennes. Ainsi, certains individus peuvent présenter un rapport T/E naturellement supérieur au seuil, conduisant à des faux positifs, tandis que d’autres peuvent prendre de faibles doses de testostérone sans dépasser ce seuil, générant des faux négatifs.
De plus, l’épitestostérone n’étant pas une substance interdite, un sportif peut en consommer afin de rééquilibrer artificiellement le rapport T/E et le maintenir en dessous du seuil de 4. D’autres facteurs peuvent également influencer ce rapport, comme la dilution des urines par une consommation excessive de liquides, ou l’utilisation de produits masquants tels que l’éthanol, la DHEA ou l’androstènedione, qui peuvent modifier le métabolisme des stéroïdes.
Ainsi, un rapport T/E supérieur à 4 ne constitue pas en soi une preuve formelle de dopage, mais un signal d’alerte. Dans ce cas, des analyses complémentaires doivent être réalisées, sauf si le profil stéroïdien de l’athlète est connu comme physiologique. Malgré ses limites, ce test est maintenu par la WADA en raison de son coût réduit, de sa simplicité et de son efficacité comme outil de criblage.
La spectrométrie de masse à rapport isotopique (IRMS)
La spectrométrie de masse à rapport isotopique (IRMS) est la méthode de référence pour confirmer l’origine exogène de la testostérone. Elle est appliquée lorsque le rapport T/E dépasse 4, mais également lorsque les concentrations absolues de testostérone et d’épitestostérone dépassent généralement 200 ng/mL, afin de détecter une éventuelle administration d’épitestostérone visant à masquer un dopage.
L’IRMS permet de mesurer avec une grande précision le rapport isotopique du carbone (¹³C/¹²C) dans les stéroïdes. Cette approche repose sur le fait que les stéroïdes synthétiques sont fabriqués à partir de précurseurs végétaux (stérols de plantes) pauvres en carbone 13, tandis que les stéroïdes endogènes produits par l’organisme humain présentent une signature isotopique différente.
Dans la méthode GC-C-IRMS (chromatographie en phase gazeuse couplée à la combustion et à l’IRMS), les stéroïdes sont d’abord séparés par GC, puis oxydés en CO₂ dans un four de combustion. Le CO₂ ainsi formé est analysé par IRMS afin de déterminer sa composition isotopique en carbone, exprimée sous forme de δ¹³C (‰) par rapport à un standard international.
Selon les critères de la WADA, la prise de stéroïdes exogènes est confirmée lorsque la valeur δ¹³C d’au moins un métabolite cible diffère d’au moins 3 ‰ de celle d’un stéroïde endogène de référence mesuré chez le même athlète. Cette approche permet une preuve directe et spécifique de l’administration de testostérone ou de ses dérivés synthétiques.
La GC-C-IRMS est beaucoup plus spécifique et fiable que le simple rapport T/E, mais elle est également longue, coûteuse et consomme une grande quantité d’urine (environ 8 à 32 mL selon le nombre de métabolites analysés). En tenant compte d’autres tests obligatoires, comme celui de l’érythropoïétine (EPO), le volume d’échantillon restant peut devenir limitant. Pour ces raisons, l’IRMS ne peut pas être utilisée en routine sur tous les échantillons et reste réservée aux cas suspects.
Détection des diurétiques
Les diurétiques sont des substances qui augmentent la production d’urine. En antidopage, ils sont utilisés comme agents masquants, car ils favorisent l’élimination rapide des substances interdites et diluent leur concentration dans l’urine, rendant leur détection plus difficile [8].
Une méthode moderne de dépistage des diurétiques et des stimulants repose sur l’utilisation de l’ultra-haute performance chromatographie liquide couplée à un spectromètre de masse haute résolution (UHPLC-HRMS). Cette approche permet un criblage large et rapide de nombreuses substances interdites au cours d’une seule analyse.
Le dépistage est réalisé en mode full scan, avec alternance des polarités positive et négative, ce qui permet la détection simultanée de plus de 120 analytes cibles. Les échantillons d’urine sont simplement dilués au dixième dans une solution contenant les étalons internes, puis directement injectés dans le système UHPLC-HRMS. Le temps d’analyse est d’environ 10 minutes par échantillon, ce qui est compatible avec une utilisation en routine.
La validation de cette méthode a montré des limites de détection comprises entre 25 et 250 ng/mL pour les diurétiques et entre 5 et 500 ng/mL pour les stimulants. Cette stratégie de criblage UHPLC-HRMS est désormais intégrée dans la lutte antidopage de routine, où elle constitue un outil puissant pour le dépistage rapide, sensible et étendu des agents masquants et des substances dopantes.
Tétracosactide (Synacthène)
Le tétracosactide est un analogue synthétique de la corticotrophine humaine (ACTH), utilisé pour stimuler la sécrétion de cortisol. Son utilisation à des fins de dopage a conduit au développement de méthodes analytiques spécifiques. Thevis et ses collaborateurs ont décrit une méthode basée sur une purification par immuno-affinité suivie d’une extraction en phase solide, puis une séparation par chromatographie liquide en phase inverse couplée à un spectromètre de masse en tandem (LC-MS/MS) [10].
Cette méthode nécessite environ 2 mL de plasma et permet d’atteindre une limite de détection d’environ 100 fmol/mL, correspondant à environ 300 pg/mL, ce qui est compatible avec les concentrations observées après administration.
Transporteurs d’oxygène à base d’hémoglobine
Les transporteurs artificiels d’oxygène à base d’hémoglobine (HBOCs) sont utilisés comme substituts sanguins et peuvent être détournés à des fins de dopage dans les sports d’endurance, afin d’augmenter le transport d’oxygène. L’un des composés les plus connus de ce groupe est Hemopure, dérivé de l’hémoglobine bovine.
L’hémoglobine bovine diffère de l’hémoglobine humaine d’environ 15 % au niveau de la séquence d’acides aminés. Après digestion trypsique, des peptides spécifiques de l’espèce bovine sont générés. Ces peptides servent de marqueurs spécifiques, qui peuvent être détectés et quantifiés par LC-MS/MS, permettant ainsi de prouver l’administration d’Hemopure chez un athlète.
Détection de l’érythropoïétine (EPO)
L’érythropoïétine (EPO) est une hormone glycoprotéique qui stimule la production de globules rouges et améliore ainsi le transport de l’oxygène, ce qui en fait une substance particulièrement attractive pour les sports d’endurance. L’EPO recombinante (rhEPO) et ses analogues (darbepoétine, CERA) sont interdits, mais leur détection est complexe car leur structure est très proche de celle de l’EPO endogène.
Les premières méthodes reposaient sur des techniques électrophorétiques (IEF et SDS-PAGE), permettant de séparer les différentes isoformes glycosylées de l’EPO en fonction de leur charge et de leur masse apparente. Aujourd’hui, ces approches sont complétées et renforcées par des méthodes LC-MS/MS, qui ciblent des peptides spécifiques après digestion enzymatique, ainsi que des marqueurs de glycosylation caractéristiques des formes recombinantes. La spectrométrie de masse permet ainsi une identification moléculaire directe de l’EPO exogène et de ses analogues, renforçant la valeur probante des contrôles antidopage.
Conclusion – Rôle de la spectrométrie de masse en antidopage
La spectrométrie de masse est devenue l’outil central de la lutte antidopage moderne, en raison de sa sensibilité, de sa spécificité et de sa capacité à analyser une très grande diversité de substances. Qu’il s’agisse de petites molécules (stéroïdes, stimulants, diurétiques), de peptides hormonaux (insuline, ACTH, EPO) ou de biomacromolécules comme les transporteurs d’oxygène à base d’hémoglobine, les techniques GC-MS/MS, LC-MS/MS, HRMS et IRMS offrent des approches complémentaires permettant à la fois le criblage, la confirmation et la preuve formelle de l’origine exogène des substances.
L’évolution des pratiques de dopage vers des molécules endogènes et des biothérapies sophistiquées a rendu les méthodes classiques insuffisantes. En réponse, la spectrométrie de masse a évolué vers des approches de plus en plus ciblées, sensibles et discriminantes, capables de différencier les composés synthétiques des substances naturellement produites par l’organisme.
Grâce à cette puissance analytique, la spectrométrie de masse ne se limite plus à un simple outil de détection, mais constitue aujourd’hui un pilier scientifique et juridique du système antidopage mondial, garantissant à la fois l’équité sportive et la protection des athlètes.
Référence
[2] R. R. Ventura, M. Roig, N. Montfort, P. Sáez, R. Bergés and J. Segura, European Journal of Mass Spectrometry 2008, 14, 191.
[3] L. Politi, L. Morini and A. Polettini, Clinica Chimica Acta 2007, 386, 46-52.
[4] F. Marclay, E. Grata, L. Perrenoud and M. Saugy, Forensic Science International 2011, 213, 73-84.
[5] K. R. Mueller, J. Grosse, R. Lang and D. Thieme, Journal of Chromatography B: Biomedical Sciences and Applications 1995, 674, 1-11.
[7] P. L. Kole, G. Venkatesh, J. Kotecha and R. Sheshala, Biomedical Chromatography 2011, 25, 199-217.
[10] Determination of Synacthen in urine for sports drug testing by means of nano-ultra-performance liquid chromatography/tandem mass spectrometry Volume 23, Issue 17 15 September 2009
Pages 2669–2674