La métabolomique et la spectrométrie de masse
 La métabolomique correspond à l’analyse globale des petites molécules présentes dans un système vivant, généralement de masse moléculaire inférieure à 1500 Da. Le terme dérive de métabolome, qui désigne l’ensemble des métabolites contenus dans un organisme, un tissu, un type cellulaire ou un fluide biologique tel que l’urine, la salive ou le plasma.
La métabolomique correspond à l’analyse globale des petites molécules présentes dans un système vivant, généralement de masse moléculaire inférieure à 1500 Da. Le terme dérive de métabolome, qui désigne l’ensemble des métabolites contenus dans un organisme, un tissu, un type cellulaire ou un fluide biologique tel que l’urine, la salive ou le plasma.
Le terme métabolite regroupe toutes les molécules de petite taille impliquées dans le métabolisme ou présentes dans l’organisme, telles que les acides aminés, sucres, alcools, phosphates de sucres, amines, acides gras, lipides polaires, hormones et vitamines, mais aussi des métabolites spécialisés comme les composés phénoliques, flavonoïdes, monoterpènes, sesquiterpènes, polykétides et alcaloïdes. Elle inclut également les xénobiotiques, tels que de nombreux médicaments, biocides et perturbateurs endocriniens.
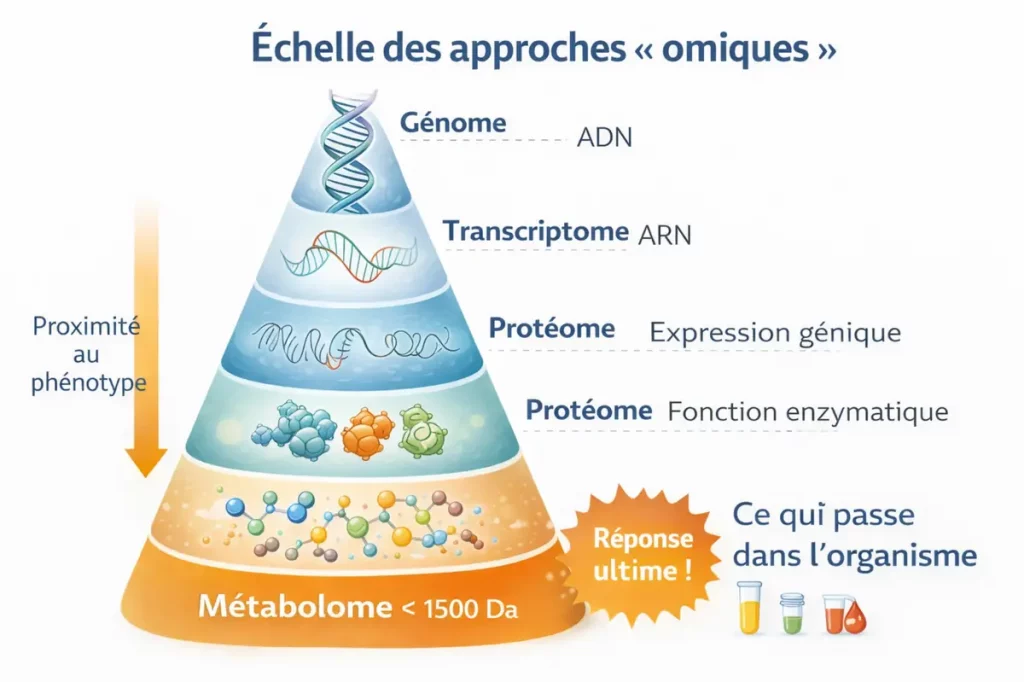
Le métabolome représente l’état final de la réponse biologique d’un organisme à des modifications génétiques, à une pathologie, à une exposition toxique ou à toute influence environnementale. Il constitue ainsi le niveau le plus proche du phénotype dans les approches « omiques » (Figure 1). Le métabolome est hautement dynamique : il varie selon les espèces, mais aussi en fonction du temps, du stade physiologique, de l’état pathologique et des conditions environnementales.
La préparation des échantillons
La préparation des échantillons est une étape clé en métabolomique, car elle doit être adaptée aux métabolites ciblés et à la matrice biologique étudiée [2]. La très grande diversité des métabolites présents dans un échantillon — en termes de polarité (hydrophiles/hydrophobes), de volatilité, de stabilité chimique et de concentration — rend impossible l’analyse exhaustive de l’ensemble du métabolome avec une seule méthode. Chez l’être humain, on estime à plus de 42 000 le nombre de métabolites, et une grande partie reste encore à identifier.
Aucune méthode de préparation ne permet de préserver simultanément tous les métabolites, car les conditions favorables à la stabilité d’un groupe de composés peuvent en dégrader d’autres ou modifier leur concentration. Il est donc nécessaire d’adapter la préparation de l’échantillon à l’objectif analytique et à la nature du prélèvement.
Pour les échantillons liquides (urine, salive), la dilution est souvent la méthode la plus simple et la plus robuste. Les échantillons sont généralement dilués dans de l’eau ultrapure ou une solution tampon, avec des rapports typiques de 1:1 à 1:10, afin de réduire les effets de matrice tout en conservant une large couverture métabolique.
Pour les tissus, l’extraction des métabolites est généralement réalisée par agitation ou homogénéisation dans des mélanges de solvants organiques, tels que chloroforme–méthanol ou chloroforme–méthanol–eau, selon des protocoles d’extraction liquide–liquide permettant de séparer les fractions polaires et lipidiques.
Les échantillons comme le plasma ou le sérum contiennent une forte proportion de protéines, qui doivent être éliminées par précipitation à l’aide de solvants organiques (acétonitrile, acétone, méthanol). Cette étape protège les systèmes LC-MS en évitant l’encrassement des colonnes et la contamination des sources d’ionisation. Toutefois, la précipitation des protéines ne permet pas d’éliminer tous les composés interférents, et certains métabolites très hydrophiles peuvent ne pas être efficacement extraits par des méthodes liquide–liquide.
Dans ces cas, l’extraction en phase solide (SPE) constitue une méthode de choix. Elle permet un nettoyage efficace de l’échantillon et la préconcentration de larges classes de métabolites, mais elle est plus longue et laborieuse, et doit être soigneusement optimisée en fonction des analytes d’intérêt.
Rôle de la LC-MS et de la HRMS en métabolomique
La spectrométrie de masse couplée à la chromatographie liquide (LC-MS) est aujourd’hui la technique la plus utilisée en métabolomique[1], en raison de sa grande sensibilité, de sa large couverture chimique et de sa compatibilité avec des molécules polaires, thermolabiles et non volatiles. Contrairement à la GC-MS, la LC-MS permet d’analyser directement un très large éventail de métabolites sans étape de dérivatisation, ce qui est essentiel pour préserver l’intégrité du métabolome.
La diversité des phases stationnaires (phase inverse, HILIC, échange d’ions, etc.) permet de séparer efficacement des métabolites de polarités très différentes, depuis les acides organiques et acides aminés jusqu’aux lipides et aux métabolites hydrophobes. Cette capacité de séparation est fondamentale en métabolomique, où des milliers de composés coexistent dans une même matrice biologique.
Le couplage à des spectromètres de masse haute résolution (HRMS), tels que les Orbitrap ou les Q-TOF, a profondément transformé la métabolomique moderne. La haute résolution et la mesure de masse exacte (souvent avec une précision < 5 ppm) permettent de déterminer des formules moléculaires, de discriminer des composés isobares et de réduire fortement les faux positifs lors de l’annotation des métabolites.
En métabolomique non ciblée, la HRMS est utilisée en mode full scan, ce qui permet de détecter simultanément des milliers de signaux correspondant à des métabolites connus ou inconnus. Ces signaux peuvent ensuite être comparés entre groupes biologiques (sujets sains vs malades, exposés vs non exposés, etc.) afin d’identifier des biomarqueurs ou des voies métaboliques perturbées.
La MS/MS haute résolution joue également un rôle clé dans l’identification structurale. Après détection d’un métabolite d’intérêt, sa fragmentation fournit un spectre de fragments qui peut être comparé à des bases de données (HMDB, METLIN, MassBank, etc.) ou interprété pour proposer une structure chimique.
Ainsi, l’association LC-HRMS constitue aujourd’hui le pilier analytique de la métabolomique, en permettant à la fois :
-
le criblage global du métabolome,
-
l’identification fiable des métabolites,
-
et la quantification relative ou absolue dans des matrices biologiques complexes.
Métabolomique ciblée et métabolomique non ciblée
La métabolomique peut être abordée selon deux stratégies complémentaires : la métabolomique ciblée et la métabolomique non ciblée.
Métabolomique non ciblée
La métabolomique non ciblée [3] vise à analyser le plus grand nombre possible de métabolites dans un échantillon biologique, sans hypothèse préalable. Elle repose principalement sur la LC-HRMS en mode full scan, qui permet de détecter simultanément des milliers de signaux correspondant à des métabolites connus ou inconnus.
Cette approche est utilisée pour :
-
découvrir de nouveaux biomarqueurs,
-
explorer des perturbations métaboliques associées à une pathologie,
-
étudier l’effet d’un médicament ou d’une exposition environnementale.
Les données générées sont ensuite traitées par des méthodes statistiques multivariées (PCA, PLS-DA, etc.) afin d’identifier les métabolites différenciellement exprimés entre groupes. Les composés d’intérêt sont ensuite annotés à l’aide de la masse exacte, du temps de rétention et des spectres MS/MS.
L’inconvénient majeur de cette approche est la complexité des données et le fait que de nombreux signaux restent non identifiés.
Métabolomique ciblée
La métabolomique ciblée se concentre sur un ensemble défini de métabolites, appartenant le plus souvent à une ou plusieurs voies biologiques connues (acides aminés, stéroïdes, acides gras, nucléotides, etc.). Elle est généralement réalisée par LC-MS/MS en mode MRM (ou SRM) sur des instruments triple quadripôle, ou par HRMS en mode ciblé.
Cette approche permet :
-
une quantification précise,
-
une excellente sensibilité,
-
une très bonne reproductibilité,
ce qui la rend particulièrement adaptée à la validation de biomarqueurs, à la recherche clinique et aux études pharmacologiques. De plus, les instruments utilisés (notamment les triple quadripôles) sont relativement robustes, peu coûteux comparés aux HRMS, et leur utilisation est bien standardisée, ce qui facilite leur déploiement dans des laboratoires non spécialisés en spectrométrie de masse.
En revanche, la métabolomique ciblée est limitée aux composés préalablement sélectionnés et ne permet pas la découverte de nouveaux métabolites, ce qui la rend moins adaptée aux approches exploratoires.
Le traitement des données métabolomiques
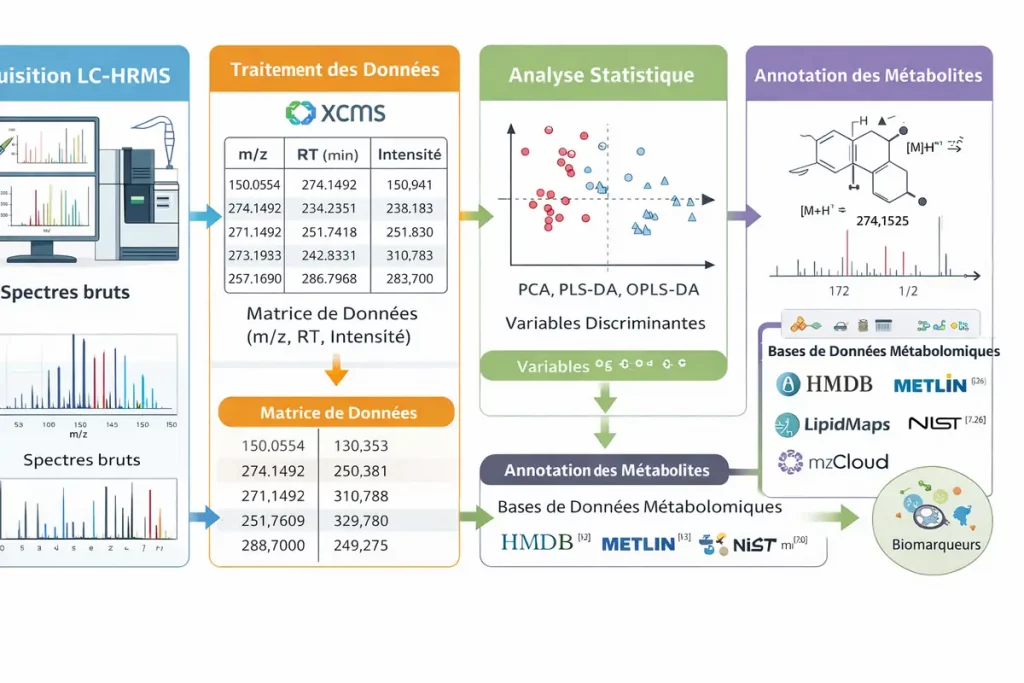 Les données générées par les différentes approches métabolomiques sont analysées à l’aide de méthodes de statistiques univariées et multivariées, qui permettent de mettre en évidence les différences entre échantillons et d’extraire l’information biologique pertinente. Les analyses métabolomiques produisent des volumes très importants de données, ce qui nécessite l’utilisation d’outils bio-informatiques spécialisés [4,5], ainsi que des compétences en statistiques, mathématiques et science des données.
Les données générées par les différentes approches métabolomiques sont analysées à l’aide de méthodes de statistiques univariées et multivariées, qui permettent de mettre en évidence les différences entre échantillons et d’extraire l’information biologique pertinente. Les analyses métabolomiques produisent des volumes très importants de données, ce qui nécessite l’utilisation d’outils bio-informatiques spécialisés [4,5], ainsi que des compétences en statistiques, mathématiques et science des données.
De nombreux logiciels de traitement existent, bien que certains soient peu connus du grand public ou développés « in house » pour des besoins spécifiques. Le principe général du traitement consiste à détecter des variables définies par un rapport m/z et un temps de rétention, puis à associer à chacune l’aire sous le pic chromatographique dans chaque échantillon, ce qui permet une quantification relative.
Parmi les outils les plus utilisés figure XCMS, qui permet la détection de pics, l’alignement des temps de rétention et la quantification relative. XCMS est intégré dans l’environnement statistique R, qui constitue une plateforme puissante pour l’analyse multivariée (PCA, PLS-DA, etc.) et l’interprétation biologique des données.
Des algorithmes spécialisés ont également été développés pour le profilage de familles spécifiques de métabolites [6,7]. Par exemple, le logiciel LipidSearch, développé par Taguchi et ses collaborateurs, utilise des schémas de fragmentation et de pertes neutres observés dans les spectres MS/MS et MS³ pour identifier des espèces lipidiques telles que les phosphatidylcholines, phosphatidylsérines et sphingomyélines.
L’identification des métabolites repose largement sur la comparaison des données expérimentales avec des bases de données. Des bases généralistes comme PubChem et ChemSpider fournissent des informations chimiques et structurales, tandis que des bibliothèques de spectres de masse telles que NIST contiennent des spectres d’ions précurseurs et de fragments.
En métabolomique, des bases spécialisées en HRMS et MS/MS sont particulièrement importantes. La base METLIN [8–10] contient des milliers de métabolites avec leurs spectres MS/MS et est interconnectée avec des ressources biologiques telles que KEGG (Kyoto Encyclopedia of Genes and Genomes), facilitant l’interprétation métabolique.
Plus récemment, la bibliothèque mzCloud [11] a été développée pour fournir des spectres de fragmentation de haute qualité et permettre l’annotation avancée des métabolites. Elle utilise notamment le concept de PIF (Precursor Ion Fingerprint) [12], qui repose sur l’analyse des motifs de fragmentation caractéristiques d’un ion précurseur. Cette approche permet de proposer des structures chimiques plausibles et d’identifier des composés inconnus, même lorsqu’ils ne sont pas présents dans la base de données.
Ainsi, le traitement des données métabolomiques combine chimie analytique, statistiques et bio-informatique afin de transformer des milliers de signaux MS en informations biologiquement interprétables.
Conclusion – La métabolomique et la spectrométrie de masse
La métabolomique offre une vision globale et fonctionnelle de l’état biologique d’un organisme. En mesurant les métabolites, elle reflète directement l’activité enzymatique, les voies métaboliques et l’interaction avec l’environnement, ce qui la place au plus près du phénotype.
La spectrométrie de masse, en particulier la LC-HRMS et la LC-MS/MS, est devenue l’outil central de la métabolomique moderne grâce à sa sensibilité, sa large couverture chimique et sa capacité à analyser des matrices biologiques complexes.
L’approche non ciblée permet la découverte de nouveaux biomarqueurs et l’exploration de mécanismes biologiques inconnus, tandis que l’approche ciblée offre une quantification fiable pour la validation clinique et le suivi thérapeutique.
Ainsi, la combinaison de la métabolomique et de la spectrométrie de masse constitue aujourd’hui un pilier de la recherche biomédicale, ouvrant la voie à une médecine de précision, fondée sur la compréhension fine des perturbations métaboliques associées aux maladies.
Référence
[3] Castrillo JI, Hayes A, Mohammed S, Gaskell SJ, Oliver SG. An optimized protocol for metabolome analysis in yeast using direct infusion electrospray mass spectrometry. Phytochemistry 2003;62:929–937
[4] Shulaev, V. Metabolomics technology and bioinformatics. Brief. Bioinform. 2006, 7, 128–139.
[5] Katajamaa, M.; Oresic, M. Data processing for mass spectrometry-based metabolomics. J Chromatogr A 2007,
1158, 318–328
[6] Taguchi, R.; Ishikawa, M. Precise and global identification of phospholipid molecular species by an Orbitrap
mass spectrometer and automated search engine Lipid Search. J. Chromatogr. A 2010, 1217, 4229–4239.
[7] Herzog, R.; Schwudke, D.; Shevchenko, A. LipidXplorer: Software for quantitative shotgun lipidomics
compatible with multiple mass spectrometry platforms. Curr. Protoc. Bioinform. 2013, 11, 14.12:1–14.12:30.
[8] Scripps Center for Metabolomics and Mass Spectrometry. Available online: https://metlin.scripps.edu
(accessed on 24 May 2016).
[10] Tautenhahn, R.; Cho, K.; Uritboonthai, W.; Zhu, Z.; Patti, G.J.; Siuzdak, G. An accelerated workflow for
untargeted metabolomics using the METLIN database. Nat. Biotechnol. 2012, 30, 826–828.
[11] Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.;
Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751.
[12] Sana, T.R.; Roark, J.C.; Li, X.; Waddell, K.; Fischer, S.M. Molecular formula and METLIN personal metabolite
database matching applied to the identification of compounds generated by LC/TOF-MS. J. Biomol. Tech.
2008, 19, 258–266.
[13] mzCloud—Advanced Mass Spectral Database. Available online: https://www.mzcloud.org (accessed on
24 May 2016).
[14] Sheldon, M.T.; Mistrik, R.; Croley, T.R. Determination of ion structures in structurally related compounds
using precursor ion fingerprinting. J. Am. Soc. Mass Spectrom. 2009, 20, 370–376