Simulation spectre MS/MS
| m/z | Int. | Annotation | m/z | Int. | Annotation | m/z | Int. | Annotation |
|---|
Simulateur spectre MS/MS peptides
Ce simulateur MS/MS peptide propose une approximation théorique de la fragmentation d’un peptide et de la distribution relative des fragments observés.
Il s’appuie sur :
-
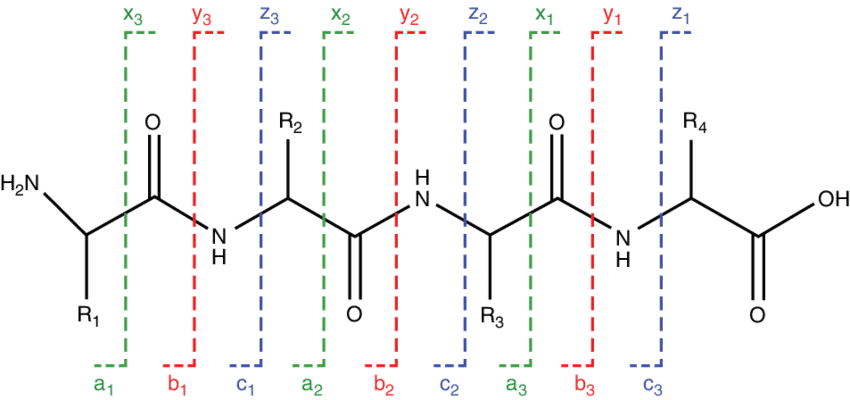
des règles classiques de fragmentation (séries b/y, pertes neutres, effet proline, etc.) ;
-
le modèle du proton mobile et des tendances liées aux affinités protoniques (la protonation influence la probabilité de rupture autour de certains résidus) ;
-
une calibration empirique inspirée de spectres MS/MS réels acquis sur un spectromètre de masse Orbitrap en mode de fragmentation HCD (Higher energy Collision Dissociation).
Limites du modèle
Même avec ces règles physico-chimiques, il est impossible de prédire exactement un spectre expérimental. La fragmentation réelle dépend de nombreux paramètres souvent non observables directement :
-
la conformation du peptide et sa dynamique en phase gazeuse ;
-
la localisation effective des charges et les réarrangements de protons ;
-
des effets à distance (résidus éloignés du site de coupure qui modifient l’énergie de dissociation) ;
-
l’énergie de collision, l’état de charge, la composition et les conditions instrumentales.
Le spectre généré doit donc être interprété comme un spectre attendu / plausible, utile pour :
-
la protéomique ciblée (SRM, PRM, DIA),
-
l’annotation des spectres MS/MS,
mais pas comme une reproduction exacte d’un spectre expérimental.
Peptides issus d’une protéine
Pour simuler des peptides générés à partir d’une protéine (trypsine, Lys-C, etc.), vous pouvez utiliser le module de digestion enzymatique intégré à ce simulateur.
Rappelle nomenclature de fragmentation peptide